
慢病毒产生和滴度
简介
根据所需的用途,sgRNA 库可以由慢病毒、逆转录病毒或腺相关病毒(AAV)提供。慢病毒和逆转录病毒整合到基因组中,而腺相关病毒不整合, 因此对于筛选,腺相关病毒的递送仅限于非分裂细胞。相反,逆转录病毒只转导分裂细胞。此外,与慢病毒和逆转录病毒相比,腺相关病毒的插入容量更小。因此,到目前为止,大多数的筛选依赖慢病毒递送。
材料与仪器
器材:离心管、Stericp 过滤器等
试剂:
① Opti-MEM I 还原血清培养基(Thermo Fisher, cat. no. 31985062)
② pMD2.G(Addgene, cat. no. 12259)
③ psPAX2(Addgene, cat. no. 12260)
④ pcDNA3-EGFP 转染控制质粒(Addgene, cat. no. 13031)
⑤ Lipofectamine 2000 转染试剂(Thermo Fisher, cat. no. 11668019)
⑥ PLUS 试齐 U(Thermo Fisher, cat. no. 11514015)
⑦ Polyethylenimine HC1 MAX, 线性,分子量 40000(PEI Max; Polysciences, cat. no. 24765-1)
⑧ 聚凝胺(Hexadimethrine bromide; Sigma-Aldrich, cat. no. 107689-10 G)
⑨ Blasticidin S HC1(Thermo Fisher, cat. no. Al 113903)
⑩ 二盐酸瞟吟霉素(Thermo Fisher, cat. no. Al 113803)
⑪ 潮霉素 B(Thermo Fisher, cat. no. 10687010)
⑫ 博来霉素(Thermo Fisher, cat. no. R25001)
⑬ CellTiter-Glo 发光细胞活力测定(Promega, cat. no. G7571)
步骤
慢病毒产生和滴度的基本过程可分为如下几步:
A. 建立抗生素杀伤曲线。在生产慢病毒和滴度之前,建议绘制用于选择 sgRNA 文库的抗生素和相关细胞系中用于筛选的其他必要成分的杀伤曲线。 为了做到这一点,种子细胞以 10% 的汇合度在含有一系列抗生素浓度的培养 基中进行筛选目。
B. 每 3 天更换一次含抗生素的培养基。4~7 天后,选择足以杀死所有细胞的最低浓度的抗生素。关键步骤:使用最低浓度的抗生素是很重要的, 以避免选择过于严格而偏向于转导多个 sgRNA 的细胞。
C. HEK 293FT 培养。在 D10 培养基中于 37 ℃, 5% 的 CO2 的培养箱中培养细胞。
D. 传代时,将培养液吸出,在 T225 瓶的侧面轻轻加入 5 mL 的 TrypLE,以免细胞脱落。去除 TrypLE, 在 37 °C 下培养 4~5 min, 直到细胞开始分离。向烧瓶中加入 10 mLD10 培养基,轻轻吹打以分离细胞,然后将细胞转移到一个 50 mL 的离心管中。关键步骤:通常每隔 1 ——2 天以 1:2 或 1:4 的比例传代细胞,绝不许细胞达到 70% 以上的融合度。对于生产慢病毒,建议 使用传代数小于 10 的 HEK 293FT 细胞。
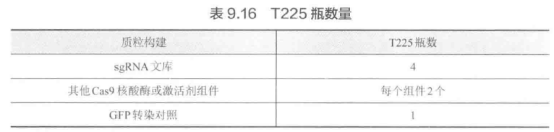
E. 准备转染细胞。转染前 20~24 小时,将分离良好的细胞接种于 T225 瓶中,每个瓶有 1.8X107 个细胞,共 45 mL D10 培养基。每个质粒构建推荐使用的 T225 瓶数量见表 9.16。
关键步骤:加入的细胞不要超过推荐密度,因为这样做可能会降低转染效率。
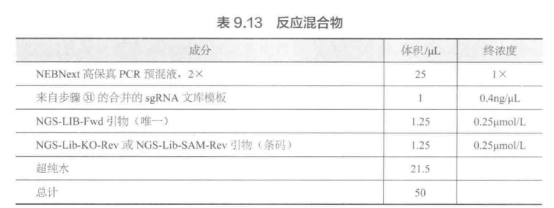
F. 慢病毒质粒转染。步骤 E 的 T225 瓶中的细胞在 80%~90% 融合度时转染效果最佳。使用 Lipofectamine 2000/PLUS 和聚乙烯亚胺(PEI)等转染慢病毒。对于每个慢病毒靶标,请将以下慢病毒靶标放入 15 mL 或 50 mL 离心管中,并相应按比例扩大(表 9.17)。

关键步骤:在推荐的细胞密度下进行转染对取得最大转染效率至关重要低密度可导致 Lipofectamine 2000 对细胞产生毒性,而高密度可降低转染效率。
G. 按照表 9.18 制备 PLUS 试剂混合物并颠倒混合。

H. 将 PLUS 试剂混合物加入慢病毒靶标混合物中,颠倒混匀,室温孵育 5 min。
I. 按表 9.19 所示制备 Lipofectamine 试剂混合,颠倒混合。

J. 将慢病毒靶标和 PLUS 试剂混合物加入 Lipofectamine 试剂混合物中,颠倒混匀,室温孵育 5 min。
K. 吸取 9 mL 来自步骤 J 的慢病毒转染混合物加入来自步骤 E 的每个 T225 瓶中,并轻典动以进行混合。将 T225 瓶放回培养箱。
L. 4 小时后,用 45 mL 预热的 D10 培养基替换原培养基。组成型 GFP 表达质粒转染对照可以指示转染效率。
M. 收集并储存慢病毒。慢病毒转染开始后 2 天,汇集用相同质粒构建体转染的 T225 瓶中的慢病毒上清液,并使用 Millipore 的 0.45 μm Stericp 过滤器滤出细胞碎片。滤过后的慢病毒上清液可以分装并保存在 -80 ℃。避免反复冻融慢病毒上清液。
N. 通过转导测定慢病毒滴度。CR1SPR-Cas9 系统已被用于许多哺乳动物 细胞系。每个细胞系的条件可能有所不同。应使用筛选过的相关细胞系来确 定慢病毒滴度。下面详细介绍 HEK 293FT 细胞(A)和 hESC HUES66 细胞(B)的转导条件和病毒滴度的计算。
1)HEK 293FT 细胞转染后的慢病毒转导和滴度。
1. HEK 293FT 培养和传代,请参阅步骤 CD。
2. 制备用于转染的细胞。对于每种慢病毒,以 3X106 的细胞密度接种 12 孔板的 6 孔,每孔 2 mL D10 培养基,其中含有 8 μg/mL 的聚凝胺。在每个孔中, 分别添加 400 μL、200 μL、100 心 50 μL、25 μL 或 0 μL 步骤 M 中的慢病毒上清液。上下吹打将各孔充分混合均匀。
3. 在 33 ℃ 条件下,1000 Xg 离心 12 孔板 2 小时对细胞进行转染,转染后 孔板送冋培养箱。
4. 转染细胞重新铺板以计算病毒滴度。转染结束后 24 小时,离心,除去 培养基,每孔用 400 μL TrypLE 轻轻冲洗,添加 100 μL TrypLE, 并在 37 ℃ 孵育 5 min 以解离细胞。每孔加入 2 mL D10 培养基,通过上下吹打以重悬细胞。
5. 使用 Cellometer 图像细胞仪针对 0 μL 慢病毒上清液条件确定细胞浓度。
6. 对于每种病毒条件,基于 100 mL D10 培养基在步骤 1)5 中确定的细胞计数,以 4X103 个细胞的密度(来自步骤 1)4)接种 96 孔透明底部黑色组织培养板中的 4 孔。再添加 100 μL D10 培养基和相应的选择抗生素。将病 毒以适当的终浓度加入 2 孔中,并将 100 μL 常规 D10 培养基加入其他 2 孔中。
7. 接种 72?96 小时后,当无病毒、无活细胞和无抗生素选择条件下细胞达到 80%~90% 融合度时,使用 CellTiter Gio 方法量化每个条件下的细胞活力。关键步骤:Cell Titer Gio 可以在 PBS 中以 1:4 稀释,降低成本的同时仍能达到最佳效果。
8. 对于每种病毒状况,将感染复数(MO1)计算为在抗生素选择条件下 2 孔的平均发光强度,或活性除以无抗生素选择的条件下 2 孔的平均发光损吋。 慢病毒上清液体积和 MOI 之间的线性关系在较低体积下有望实现,而在较高体积下则会达到饱和。
2)通过混合检测 hESC HUES66 细胞的慢病毒转导和滴度。
1. HUES66 细胞培养。HUES66 细胞(hESC 细胞系)常规在无培养层的 情况下培养,并在 GelTrex 涂层的组织培养板上使用 mTeSRl 培养基。要覆盖 100 mm 的组织培养皿,在 5 mL 的冷 DMEM 中以 1 : 100 稀释冷的 GelTrex, 覆盖住整个培养皿表面,然后将"泞『 37 °C 的培养箱中至少 30 min。铺板而, 吸出 GelTrex 混合物。在传代和铺板过程中,在 mTeSRl 培养基中进一步补充 10 μmol/L ROCK 抑制剂。每天更换 mTeSRl 培养基。
2. HUES 66 传代。吸出培养基并在 100 mm 组织培养皿的侧面轻轻加入 10 mL DPBS 冲洗一次,避免细胞脱落。加入 2 mL 的细胞消化液解离细胞,并 在 37 °C 下孵育 3~5 min, 直至细胞解离。加入 10 mL DMEM 重悬解离的细胞, 并以 200 Xg, 5 min 沉淀细胞。除去上清液,将细胞重悬于含 10 μmol/L ROCK 抑制剂的 mTeSRl 培养基中,然后将细胞重铺在 GelTrex 包被的板上。接种后 24 小时更换普通的 mTeSRl 培养基。关键步骤:近常以 1:5 或 1:10 的比例, 每 4~5 天传代一次细胞,绝不允许细胞达到 70% 以上的融合度。
3. 制备用于慢病毒转导的细胞。对于每种慢病毒,以 5X105 个细胞的密 度加入 Geltrex 包被的
4. 孔板进行培养,每孔 2 mL mTeSRl 培养基。在每个 孔中,加入 400 μL、200 μL、100 μL、50 μL、25 μL 或 0 μL 慢病毒上清液,用 DPBS 补充至 3 mL 的总体积,并补充 10 pmol/L ROCK 抑制剂。在没有病毒的 情况下,以相同的接种密度在另一个无抗生素选择的对照平板上接种。通过 上下吹打将各孔充分混匀。
5. 慢病毒转导 24 小时后,用含有相关选择性抗生素的 mTeSRl 培养基更 换培养基。对于无抗生素选择的对照孔,用正常 mTeSRl 培养基更换。每天 使用含或不含选择抗生素的 mTeSRl 培养基更换,直到培养皿准备充分可以 进行下一步。
6. 病毒滴度的计算。开始使用选择抗生素后 72~96 小时,当无病毒条 件下没有活细胞和无抗生素选择的控制条件下达到 80%~90% 融合时,用 2 mL DPBS 冲洗细胞,加入 500 μL 细胞消化液,然后在 37 °C 下 3~5 min 孵 育以解离细胞。加入 2 mL DMEM 并充分混合。
7. 使用 Cellometer 图像细胞仪计数并记录每个孔中的细胞数量。
8. 对于每种病毒条件,将 MOI 计算为抗生素选择条件下的细胞数除以无 抗生素选择对照下的细胞数。慢病毒上清液体积和 MOI 之间的线性关系在较 低体积下有望实现,而在较高体积下则会达到饱和。


相关文章

