
建立随机重叠 DNA 插入文库实验
材料与仪器乙酸铵 ATP 乙醇 甘油 蒸馏水 IPTG MgCl2 NaCl 酚 聚乙二醇 TE TM 缓冲液 Tris-HCl TTE 缓冲液 X-gal T
材料与仪器
乙酸铵 ATP 乙醇 甘油 蒸馏水 IPTG MgCl2 NaCl 酚 聚乙二醇 TE TM 缓冲液 Tris-HCl TTE 缓冲液 X-gal T4 噬菌体 DNA 连接酶 T4 噬菌体 DNA 聚合酶 T4 噬菌体多核苷酸激酶 大肠杆菌 DNA 聚合酶 琼脂糖凝胶 聚丙烯酰胺凝胶 噬菌体 DNA DNA 分子质量标准 dNTP 溶液 靶 DNA LB 或YT 培养基
离心机和转子 深孔微量板、盖、帽 微量滴定板 多头移液器 多管涡旋振荡混合器 银色胶带 超声仪或喷霁器 牙签 水浴 大肠杆菌感受态细胞 M13噬菌体载体 DNA
离心机和转子 深孔微量板、盖、帽 微量滴定板 多头移液器 多管涡旋振荡混合器 银色胶带 超声仪或喷霁器 牙签 水浴 大肠杆菌感受态细胞 M13噬菌体载体 DNA
步骤
材料
缓冲液和溶液
乙酸铵(10mol/L)
ATP
乙醇
甘油(无菌,100%)
水(去离子蒸馏水)
IPTG
MgCl2
NaCl
酚(pH7.6 Tris 饱和)
酚:氯仿(1:1,m/V)
聚乙二醇(30% m/V PEG 8000) 溶于 2.5mol/LNaCl
可选用,参见步骤 1
聚乙二醇(20% m/V PEG 8000) 溶于 2.5mol/LNaCl
TE(pH7.6)
10XTM 缓冲液
0.5mol/LTris-HCl(pH8.0)
150 mmol/LMgCl2
TTE 缓冲液
含 0.5%Triton X-100TE(pH8.0)
X-gal
酶和缓冲液
T4 噬菌体 DNA 连接酶
T4 噬菌体 DNA 聚合酶
T4 噬菌体多核苷酸激酶
大肠杆菌 DNA 聚合酶 I Klenow 片段
凝胶
琼脂糖凝胶(两种 1% 和一种 0.7%)
聚丙烯酰胺凝胶(中性,5% 聚丙烯酰胺)或琼脂糖凝胶(0.8% 低熔点琼脂糖)
核酸和寡核苷酸
λ噬菌体 DNA 或Φ174DNA 用 Alu I[1ug Alu I, 溶于 20ul TE(pH7.6)]完全酶解]。
酶解后,用等体积酚: 氯仿抽提 DNA, 去除限制性内切核苷酶。乙醇沉淀 DNA 重溶于 TE(PH7.6) 中,按步骤 13 測试酶解 DNA 与去磷酸化、线性化栽体的连接效率。如采使用商品化的栽体就不需要消化 DNA 了。
DNA 分子质量标准
适当大小的 DNA 分子质量标准,如 Life Technologies 公司用 Sau3AI 消化的 PUC18pUC19DNA, 或相差 123bp 的梯度分子质量标准;New England Biolabs 用 Hae Ⅲ消化的ΦX174 DNA。
dNTP 溶液,含有 4 种 dNTP, 每种浓度为 2.0 mmol/L
靶 DNA
靶 DNA 制备通常由用限制酶消化高容量栽体(如:黏粒栽体、BAC、PAC 或 P1) 构建的重组体获得。所用限制酶的酶切位点应在克隆片段的序列中不存在。如果可能尽量使用能产生黏性末端的限制性酶,这可以简化纯化的粑 DNA 的连接(步骤 1)。靶 DNA 片段从载体中酶切下来后,用低熔点琼脂糖凝胶电泳纯化(请参见第 5 章的方案 6 或 7)。用 TE(pH7.6) 溶解纯化的 DNA,浓度为取 DNA 溶液(50ng),用琼脂糖凝胶电泳检査 DNA 的完整性和回收率。
培养基
LB 或含有 5 mmol/L MgCl2 的 2xYT 培养基
顶层琼脂糖
YT琼脂平板
离心机和转子
转速3000r/min离心深孔微量板和标准微量滴定板的离心机。
例如配备微孔板架的贝克曼GPR离心机,配备摆式转头(M4型)和微孔板架的JocanCR422型离心机。
专用设备
深孔微量板、盖、帽
微孔要能容纳1~1.2 ml, 并能经受住3000r/min的离心。如96孔平底板(DBM Scientific,San Fernando,California)和盖子(V3-Verl-Tips-Cover,Ulater Sciemifici购自Baxter Inc.,McGaw Park,Illinois)。此外,还有配有单片盖和帽的贝克曼96管盒。制备96个M13噬菌体模板需要两盒配有盖和帽的管。
微量滴定板(96孔,配有合适的盖)
参见信息栏有关微量滴定板部分。
多头移液器(8或12个头)
多管涡旋振荡混合器
银色胶带(3M公司产品)或相应物品
胶带必须确保96管盒密封不漏水。在美国和墨西哥R.S.Hughes公司(http://www.rshughes.com)提供3M公司的银色胶带。
超声仪或喷霁器
关于超声,可以用微型探头超声仪或杯-角型超声仪。本方案中推荐使用杯-角型超声仪(如配备CL4杯-角型探头的XL2015型超声仪,Heat Systems产品),有两点理由:1.DNA置于微量离心管中,体积通常只有20--25ul;2. 因为杯-角杯型超声仪探头不接触DNA溶液,没有污染。关于喷雾器,对一次性的治疗用喷雾器进行改造的方法见附录8。
牙签
可调节16°C、37°C、68°C和80°C的水浴。
载体和菌株
M13噬菌体载体 DNA,包括蓝白筛选、线性化、平末端和去磷酸化的载体DNA。
这些种类的预制栽体DNA可以从公司购得,也可以按照本方案后面提供的附加方法制备。
适当菌株的大肠杆菌感受态细胞(如XLIF'-Blue、DH5aF')
高效的大扬杆菌感受态细胞(>109 转染子/ug 闭环栽体DNA) 对获得高丰度的靶 DNA 库是十分重要的。预制备的高效大肠杆菌感受态细胞可以从公司购得,也可参照第 1 章的方法 23~26 制备。如果使用电转移仪,我们推荐使用电转大肠杆菌感受态细胞,可以获得高效转化。
方法
靶 DNA 自身连接
自身连接是确保耙 DNA 末端序列在构建文库的片段中有足够的量所需要的。
1. 将 5~10 g 纯化的靶 DMA 加入一只干净的离心管中,并加入:
10XT4 噬菌体 DNA 连接酶缓冲液 2.5ul
5 mmol/LATP 2.5ul
30%m/V PEG8000(可选加) 5.0ul
T4 噬菌体 DNA 连接酶 0.5~2.0Weiss 单位
H20 至终体积 25ul
混合液于 16°C 温育 4 h, 然后于 68°C 加热 15 min 使连接酶失活。
如果靶 DNA 是平末端,在反应中加入 PEG 可以提髙连接效率,黏末端连接,连接酶用量为 0.5Weiss 单位;平末端连接,连接酶用量为 2.0Weiss 单位。
有些商品连接酶缓冲液中含有 ATP, 此时无须添加 ATP。
2. 加入 175ul TE(pH7.6), 用酚: 氯仿抽提以纯化连接好的 DNA。用 2mol/L 乙酸铵和 3 倍体积乙醇沉淀 DNA,用微量离心机以最大转速离心 5 min, 回收 DNA。于室温用 0.5 ml70% 乙醇洗涤沉淀,再次离心。
3. 尽可能去除上清液,在室温下使最后残留的痕量乙醇挥发殆尽。在微量离心管中加入 25ul TE(pH7.6) 溶解 DNA。
靶 DNA 的断裂
下列方法是 Birren 等(1997) 方法的改进。
4. 用超声处理或喷雾处理使粑 DNA 断裂为 0.8~1.5kb 长的片段(参见表 12-1)。
超声断裂 DNA(参见图 12-3)
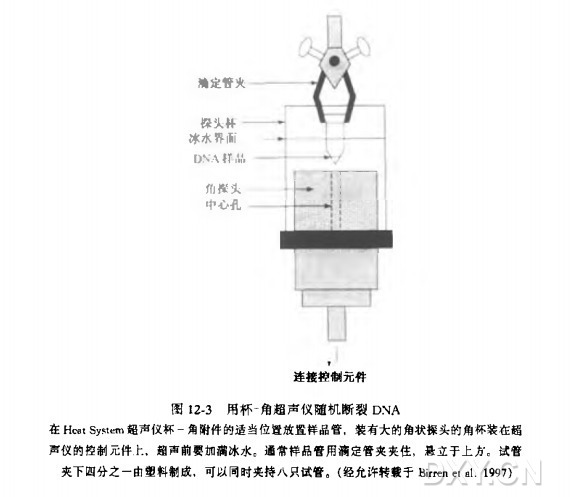
a. 在超声仪的角杯中注满冰水,打开超声仪开关. 设置好时间,功率设为 10, 用两次 40 秒脉冲预热超声仪。
冰水可以防止 DNA 变性,超声不同样品前最好更换角杯中的冰水。
b. 将装有 DNA 的离心管放置于冰水中,离心管的底部恰好高于角杯探头中心孔 1~2 mm,超声处理 DNA(参见图 12-3)。
通过超声处理试验样品 DNA, 并用步骤 4 描述的方法分析超声结果,确定最适超声处理条件。对于大多数 DNA 样品,功率设为 3, 两次 6 秒脉冲主要产生 500~2000bp 大小的片段。
c. 快速离心使超声处理的 DNA 溶液沉积于离心管底部,并置于冰上。
d. 取 lul 超声处理的 DNA 样品和适当大小的 DNA 分子质量标准,用 0.7% 琼脂糖凝胶电泳分析 DNA 片段大小。电泳时余下的 DNA 样品仍放置在冰上。
如果得到的 DNA 片段大小不合适,改变超声条件,再次进行超声处理。如果得到的 DNA 片段大小合适,继续按步骤 5 所述进行 DMA 末端修补。
喷雾断裂 DNA(请参见图 12-4)

a. 准备如下 DNA 溶液,并置于喷雾杯中。
DNA 样品(来源于步骤 3 ) 5~10ug
10XTM 缓冲液(pH8.0) 200ul
无菌 100% 甘油 1 ml
无菌水 至终体积为 2 ml
b. 将 DNA 样品置于冰浴中,依据经验用最适条件喷雾处理 DNA 样品。
DNA 片段的大小主要决定于氮气的压力。例如,8psi(0.56 kg/cm2) 压力处理黏粒栽体 DNA 主要产生 1000~2500bp 大小的片段。对一个新的靶 DNA 的最适喷雾压力和喷雾时间要依据经验确定。同超声处理-样,冰水浴可以防止 DNA 降解,同时对产生大小均匀的 DNA 片段有重要作用。
C. 将喷雾器置于合适的离心机转头中,用聚苯乙烯泡沫塑料做衬蛰。于 4°C 2000 g(配备微孔板放置装置的离心机用 1000r/min) 快速离心使样品 DNA 溶液沉积于喷雾杯的底部。
d. 将 DNA 样品溶液分成四份,转入 1.5 ml 离心管中,按标准方法乙醇沉淀 DNA, 真空干燥 DNA 沉淀。
e. 用 35ul TE(pH7.6) 溶解每一份 DNA 沉淀,取 1ul 喷雾处理的 DNA 样品和适当分子质量大小的 DNA 分子质量标准,用 0.7% 琼脂糖凝胶电泳分析 DNA 片段大小。电泳时余下的 DNA 样品 4°C 保存。
如果得到的 DMA 片段大小不合适,改变喷雾条件,再次进行喷雾处理。如果得到的 DNA 片段大小合适,继续按步骤 5 所述进行 DNA 末端修补。
DNA 的修补、磷酸化和大小选择
喷雾处理和超声处理产生的 DMA 末端是高度不均一的,包括平末端和残损末端、带有磷酸基团和不带磷酸基团的末端。因为在这些分子中只有一部分能被 DNA 聚合酶修补,流体静力剪切的 DNA 克隆到 M13 噬菌体上的效率通常较低。不过超声处理 5~10ug 靶 DNA, 经过修补和选择大小适当的片段,一般能形成几千个重组克隆。
5. 在打断的 DNA(约 25ul) 中,加入:
10XT4 噬菌体 DNA 聚合酶缓冲液 4.0ul
2.0 mmol/L 含四种 dNTP 溶液 4.0ul
T4 噬菌体 FJNA 聚合酶 5 单位
水 至终体积 40ul
于室温温育 15 min,然后加入约 5 单位 Klenow 片段,继续于室温温育 15 min。
该反应使用两种 DNA 聚合酶修补由于流体静力剪切产生的 DNA 片段的残损末端。T4 噬菌体 DNA 聚合酶补平 3'凹端,并且其外切酶活性可去除突出端。Klenow 片段提供了第二种补平 3'凹端的工具. 有关内容可参见信息栏"大肠杆菌 DNA 聚合酶 Ⅰ Klenow 片段"部分。
6. 用酚: 氯仿抽提纯化 DNA。上层水相转移到一个干净试管中,使 NaCl 浓度达到 0.1mol/L, 加 2 倍体积乙醇,回收沉淀 DNA。用 70% 乙醇洗 DNA 沉淀。
7. 加入 25ul TE(pH7.6) 重新溶解沉淀 DNA。
8. 在微量离心管中混合如下组分:
打断的 DNA 23ul
10x 多核苷酸激酶缓冲液 3ul
20 mmol/L.ATP 3ul
T4 噬菌体多核苷酸激酶 1 单位
T4 噬菌体多核苷酸激酶催化平末端 DNA5'端磷酸化,此步骤不是必需的. 但是一般情況下可以提高 DNA 片段与载体的连接效率。
9. 于 37°C 温育 30 min。
10. 用 0.8% 低熔点琼脂糖凝胶电泳或 5% 中性聚丙烯酰胺凝胶电泳(参见第 5 章)纯化所需大小(0.8~1.5kb)DNA 片段。
为最大限度地减少污染的可能性,应在断裂靶 DNA 和标准参照物之间空出几个泳道,这一点在使用平端 DNA 作为参照物时尤为重要,因其与载体 DNA 的连接效率明显高于剪切的靶 DNA。
11. 用第 5 章描述的方法从凝胶上回收靶 DNA。用 25ulTE(pH7.6) 溶解纯化的 DNA。
12. 取 1ul DNA 用 1% 琼脂糖凝胶电泳分析所纯化 DNA 的完整性和回收率。
与载体 DNA 连接
13. 设立一系列试验连接反应,其中含有 50ng(~0.01pmol) 线性化和去磷酸化载体
DNA 及浓度递增的平端、磷酸化的靶 DNA 片段(见表 12-2)。

14. 连接液通过电转移或转化适当菌株的大肠杆菌感受态细胞(参见第1章的方法23~26)。将细菌铺于含IPTG和X-gal的培养基上,37°C培养过夜。
这一步骤的目的在于找出一个靶DNA片段的浓度,以便最大限度地减小含有人为融合的靶片段的重组体,而后者会使测序更为复杂。因此,当确定大量的连接反应时(步骤16), 必规注意避免使用饱和量的靶DNA。相反,所定出的靶DNA量应为重组克隆的数目与背最相比有中等程度的提高(大约提高至5倍)。
15. 次日计数蓝噬菌斑和白噬菌斑的数目。
采用断裂的平端靶DNA所得的重组体数目大约仅为采用限制性内切核酸酶制备的平端DNA所得数目的十分之一{例如同用Alu I消化的λDNA或ΦX174DNA作为起始连接反应相比)。
16. 用最小量的断裂的平端靶DNA设立一个较大规模的连接反应,以便获得足够的重组克隆完成测序任务。用连接的 DNA转化大肠杆菌,37°C培养过夜。
这一步骤的目的在于确保毎个靶片段至少获得五个重组充隆。图12-5表明了覆盖95% 的双链靶DNA序列所必须测定的克隆的大概数目。
17, 次日收集平板,将转化子保存在适当条件下备用。从一系列单个无色噬菌斑中制备DNA 模板的方法见第 3 章的方法4。
M13重组噬菌体噬菌斑应尽快挑出和扩增(参见第 3 章的方案 2的「噬菌斑的挑取」专栏)。因为噬菌体顆粒可以在顶层琼脂中扩散至较远距离,在37°C生长长时间(>12~16 小时)的噬菌斑或者在 4°C 存放数天的噬菌斑往往已被污染。因此使用从时间较久的噬菌斑制备 DNA 进行测序反应时,背景带的强度和数目都有所增加。
用 96 管盒培养 M13 噬菌体重组克隆
按第3章的方案2 所述方法挑取单一无色噬菌斑,接种在含有 2 ml 细菌培养基的 15 ml 试管中进行培养。然后分别回收备重组病毒颗粒,纯化 DNA(参见第3章的方案 3 和 4)。由于每次只能处理 12 或 24 个克隆,模板纯化过程费时且繁琐。大规模的 DNA 测序需要成千上万的 DNA 模板,但采用有机溶剂抽提和多步离心方法不能快速、经济地制备单链 DNA 模板。取而代之的 M13 噬菌体模板大量制备方法通常包括噬菌体颗粒的纯化或结合自动化技术设备(e.g.,please see Mardis and Roe1989;Smith et al.1990) 用过滤法(Eperon1996)、磁珠法(Alderton et al.1992;Wahlberg et al.1992;Hawkins et al.1994) 或顺磁球法(Fryetal.1992;Wilson1993) 纯化单链 DNA。但是这些设备和操作设备所需的工作人员是一般的学术机构所不具备的。不过 Zollo 和 Chen(1994) 报告了一种用于在 96 孔板中培养 M13 噬菌体克隆,并制备单链 DNA 的有效的可重复的方法,满足了基于荧光的自动化 DNA 测序仪对大量模板的需求。

在鸟枪法测序中,使用一次性平底孔板或一次性试管组盒,以少 M 细菌培养物培养 M13 噬菌体重组子,同时处理 96 个样品是非常有效的。所有单链 DNA 模板制备有关的后续步骤都可以在同一块板中进行,减少了一步又一步转移克隆的工作量,使一个研究人员可在一天内制备 960 个或更多的DNA模板(Pleasesee Zollo and Chen1994)。
18. 对 96 个克隆,每一个克隆都逐一地将大肠杆菌 F'菌株(如 XLl-Blue、XLl-Blue MRF'或 HD5αF') 的单一菌落接种到 100 mlLB 或 2 x YT 培养基,并加至容量为 500 ml 的培养瓶中。于 37°C 以 300r/min 振荡培养 6~8 h。
19. 在培养液中加入 MgS04 至终浓度为 5 mmol/L。
为了保持离心时的平衡和对称,培养的 M13 噬菌体的克隆数为 96 的偶数倍。
Mg2+能提高 M13 噬菌体产量,并降低不同克隆间生长速度差异。
毎组 96 个 M13 噬菌体克隆约需要 80 ml 培养细胞。
20. 用多道移液器转移 0.8 ml 细胞培养液至 96 管盒的各管中。
21. 带上手套,用无菌牙签在每一个 96 管盒的各管中接种单一无色的 M13 噬菌斑。用牙签穿刺噬菌斑的中央,然后投入培养管中。
22. 为避免造成混乱,将牙签保留在培养管中直到所有 96 个培养管都接种完毕。
23. 当最后一个噬菌斑挑取完成,取出所有牙签,封闭培养管盒。培养管盒做好标记,放入 37°C 摇床中,以 250~300r/min 振荡培养 8~12 h。如果需要可重复步骤 20 和 21。
如果培养时间超过 12 h, 制备的单链模板将会被大量的 M13 噬菌体双链复制型 DNA 和/或染色体 DNA 所污染。来源于细菌裂解产生的 DNA 将加大双脱氧测序反应中错配引物的机会。长时间培养也将加大 DNA 克隆片段缺失和重排的机会。因此,适当短时间培养是本方法成功的关键。
M13DNA 的纯化
24. 从培养箱中取出培养管盒,2400 g(带有微量板套筒的离心机转速为 3000~3250r/min) 离心 20 min 沉淀菌体。
M13 噬菌体克隆的主要保存方法是在 50ul 悬液中加人 25ul80% 的甘油,用衡量加样器上下吹打混合溶液,贮存板保存于-80°C。如果需要再制备模板 DNA,这些板作为感染用的噬菌体来源。本方案最后要强调的是安全地保存噬菌体,在本方案中各管间有较大的交叉污染的可能,这对 DNA 测序影响不大,但对于种源的保存是不能接受的。
25. 用多道移液器在新的 96 管盒的各管中加入 120ul 含 20%PEG8000 的 2.5 ml/L NaCl 溶液。
26. 从离心机小心取出离心管,用多道移液器从各管中分别吸取 0.6 ml 上清液加入到已加有 PEG/NaCl 溶液的各管中。
重要:在本步骤中一定不要搅动菌体沉淀,混有菌体将显著降低 DNA 测序质量。
27. 用 96 管盖盖上装有噬菌体悬液和 FEG/NaCl 溶液的试管,确保形成牢固的液封,颠倒试管几次,以混合内容物。试管盒室温放置 30 min, 然后冰浴 30~60 min。
28.2400 g(带有微量板套筒的离心机转速为 3000~3250r/min) 离心 30 min, 收集沉淀噬菌体。取出各排试管,倒置于水槽上空干,在每支试管底部可见少量白色沉淀。将试管放回试管盒。
29. 当所有试管被空干,将试管盒倒置在吸水纸上几分钟空干残存的痕量液体。重新换上新吸水纸,将倒置的试管盒与吸水纸放入离心机中,以 300r/min 离心 3~5 min 去除最后残存的痕量 PEG/NaCl 溶液。
30. 从离心机中取出试管盒,检查试管底部是否仍有噬菌体沉淀。每管加入 20ul TTE 缓冲液。
如果唯菌体生长良好,沉淀呈白色不透明状;如果生长不好,沉淀呈带点蓝色不透明状。如果沉淀呈褐色团块,很可能是因为在步骤 26 取菌体量过多。棕色沉淀制备的模板测序所得结果欠佳。
31. 用 3M 银色胶带密封试管口,在多管旋涡振荡器上剧烈振荡试管盒 15~30 min。
32. 快速离心试管盒使液体沉于管底,去掉每一个 96 孔管盒底座,将试管置于 80°C 水浴中保温 10 min。
该步骤是为了裂解 M13 噬菌体颗粒。去掉管盒的底座可以确保所有的试管在 80°C 保温达到 10 min。
33. 从水浴中取出试管并冷却至室温。重新安上试管盒底座,快速离心试管盒使液体沉于管底。
34. 用多道移液器将 70ul 无菌水加到 96 孔板的各孔中,将步骤 33 获得的噬菌体裂解液转移到 96 孔板中,用移液器吹打混匀两种溶液。用一条 3M 银胶带密封 96 孔板,如果在 24~48 h 内进行测序也可以用 96 孔板盖板盖住。标记好各块板,贮存于-20°C。
每进行一次培养可以提取到 2.5~5ulM13 噬菌体单链 DNA。
35. 随机从一些孔中取 DNA 样品(5ul), 用 1% 琼脂糖凝较电泳检査 DNA 量。
如果一切正常,各管中的 DNA 产虽差别很小。DNA 循环测序反应一般需制备的 DNA 量为 2ul~7.5ul
(参见方案 6)。

缓冲液和溶液
乙酸铵(10mol/L)
ATP
乙醇
甘油(无菌,100%)
水(去离子蒸馏水)
IPTG
MgCl2
NaCl
酚(pH7.6 Tris 饱和)
酚:氯仿(1:1,m/V)
聚乙二醇(30% m/V PEG 8000) 溶于 2.5mol/LNaCl
可选用,参见步骤 1
聚乙二醇(20% m/V PEG 8000) 溶于 2.5mol/LNaCl
TE(pH7.6)
10XTM 缓冲液
0.5mol/LTris-HCl(pH8.0)
150 mmol/LMgCl2
TTE 缓冲液
含 0.5%Triton X-100TE(pH8.0)
X-gal
酶和缓冲液
T4 噬菌体 DNA 连接酶
T4 噬菌体 DNA 聚合酶
T4 噬菌体多核苷酸激酶
大肠杆菌 DNA 聚合酶 I Klenow 片段
凝胶
琼脂糖凝胶(两种 1% 和一种 0.7%)
聚丙烯酰胺凝胶(中性,5% 聚丙烯酰胺)或琼脂糖凝胶(0.8% 低熔点琼脂糖)
核酸和寡核苷酸
λ噬菌体 DNA 或Φ174DNA 用 Alu I[1ug Alu I, 溶于 20ul TE(pH7.6)]完全酶解]。
酶解后,用等体积酚: 氯仿抽提 DNA, 去除限制性内切核苷酶。乙醇沉淀 DNA 重溶于 TE(PH7.6) 中,按步骤 13 測试酶解 DNA 与去磷酸化、线性化栽体的连接效率。如采使用商品化的栽体就不需要消化 DNA 了。
DNA 分子质量标准
适当大小的 DNA 分子质量标准,如 Life Technologies 公司用 Sau3AI 消化的 PUC18pUC19DNA, 或相差 123bp 的梯度分子质量标准;New England Biolabs 用 Hae Ⅲ消化的ΦX174 DNA。
dNTP 溶液,含有 4 种 dNTP, 每种浓度为 2.0 mmol/L
靶 DNA
靶 DNA 制备通常由用限制酶消化高容量栽体(如:黏粒栽体、BAC、PAC 或 P1) 构建的重组体获得。所用限制酶的酶切位点应在克隆片段的序列中不存在。如果可能尽量使用能产生黏性末端的限制性酶,这可以简化纯化的粑 DNA 的连接(步骤 1)。靶 DNA 片段从载体中酶切下来后,用低熔点琼脂糖凝胶电泳纯化(请参见第 5 章的方案 6 或 7)。用 TE(pH7.6) 溶解纯化的 DNA,浓度为取 DNA 溶液(50ng),用琼脂糖凝胶电泳检査 DNA 的完整性和回收率。
培养基
LB 或含有 5 mmol/L MgCl2 的 2xYT 培养基
顶层琼脂糖
YT琼脂平板
离心机和转子
转速3000r/min离心深孔微量板和标准微量滴定板的离心机。
例如配备微孔板架的贝克曼GPR离心机,配备摆式转头(M4型)和微孔板架的JocanCR422型离心机。
专用设备
深孔微量板、盖、帽
微孔要能容纳1~1.2 ml, 并能经受住3000r/min的离心。如96孔平底板(DBM Scientific,San Fernando,California)和盖子(V3-Verl-Tips-Cover,Ulater Sciemifici购自Baxter Inc.,McGaw Park,Illinois)。此外,还有配有单片盖和帽的贝克曼96管盒。制备96个M13噬菌体模板需要两盒配有盖和帽的管。
微量滴定板(96孔,配有合适的盖)
参见信息栏有关微量滴定板部分。
多头移液器(8或12个头)
多管涡旋振荡混合器
银色胶带(3M公司产品)或相应物品
胶带必须确保96管盒密封不漏水。在美国和墨西哥R.S.Hughes公司(http://www.rshughes.com)提供3M公司的银色胶带。
超声仪或喷霁器
关于超声,可以用微型探头超声仪或杯-角型超声仪。本方案中推荐使用杯-角型超声仪(如配备CL4杯-角型探头的XL2015型超声仪,Heat Systems产品),有两点理由:1.DNA置于微量离心管中,体积通常只有20--25ul;2. 因为杯-角杯型超声仪探头不接触DNA溶液,没有污染。关于喷雾器,对一次性的治疗用喷雾器进行改造的方法见附录8。
牙签
可调节16°C、37°C、68°C和80°C的水浴。
载体和菌株
M13噬菌体载体 DNA,包括蓝白筛选、线性化、平末端和去磷酸化的载体DNA。
这些种类的预制栽体DNA可以从公司购得,也可以按照本方案后面提供的附加方法制备。
适当菌株的大肠杆菌感受态细胞(如XLIF'-Blue、DH5aF')
高效的大扬杆菌感受态细胞(>109 转染子/ug 闭环栽体DNA) 对获得高丰度的靶 DNA 库是十分重要的。预制备的高效大肠杆菌感受态细胞可以从公司购得,也可参照第 1 章的方法 23~26 制备。如果使用电转移仪,我们推荐使用电转大肠杆菌感受态细胞,可以获得高效转化。
方法
靶 DNA 自身连接
自身连接是确保耙 DNA 末端序列在构建文库的片段中有足够的量所需要的。
1. 将 5~10 g 纯化的靶 DMA 加入一只干净的离心管中,并加入:
10XT4 噬菌体 DNA 连接酶缓冲液 2.5ul
5 mmol/LATP 2.5ul
30%m/V PEG8000(可选加) 5.0ul
T4 噬菌体 DNA 连接酶 0.5~2.0Weiss 单位
H20 至终体积 25ul
混合液于 16°C 温育 4 h, 然后于 68°C 加热 15 min 使连接酶失活。
如果靶 DNA 是平末端,在反应中加入 PEG 可以提髙连接效率,黏末端连接,连接酶用量为 0.5Weiss 单位;平末端连接,连接酶用量为 2.0Weiss 单位。
有些商品连接酶缓冲液中含有 ATP, 此时无须添加 ATP。
2. 加入 175ul TE(pH7.6), 用酚: 氯仿抽提以纯化连接好的 DNA。用 2mol/L 乙酸铵和 3 倍体积乙醇沉淀 DNA,用微量离心机以最大转速离心 5 min, 回收 DNA。于室温用 0.5 ml70% 乙醇洗涤沉淀,再次离心。
3. 尽可能去除上清液,在室温下使最后残留的痕量乙醇挥发殆尽。在微量离心管中加入 25ul TE(pH7.6) 溶解 DNA。
靶 DNA 的断裂
下列方法是 Birren 等(1997) 方法的改进。
4. 用超声处理或喷雾处理使粑 DNA 断裂为 0.8~1.5kb 长的片段(参见表 12-1)。
超声断裂 DNA(参见图 12-3)
a. 在超声仪的角杯中注满冰水,打开超声仪开关. 设置好时间,功率设为 10, 用两次 40 秒脉冲预热超声仪。
冰水可以防止 DNA 变性,超声不同样品前最好更换角杯中的冰水。
b. 将装有 DNA 的离心管放置于冰水中,离心管的底部恰好高于角杯探头中心孔 1~2 mm,超声处理 DNA(参见图 12-3)。
通过超声处理试验样品 DNA, 并用步骤 4 描述的方法分析超声结果,确定最适超声处理条件。对于大多数 DNA 样品,功率设为 3, 两次 6 秒脉冲主要产生 500~2000bp 大小的片段。
c. 快速离心使超声处理的 DNA 溶液沉积于离心管底部,并置于冰上。
d. 取 lul 超声处理的 DNA 样品和适当大小的 DNA 分子质量标准,用 0.7% 琼脂糖凝胶电泳分析 DNA 片段大小。电泳时余下的 DNA 样品仍放置在冰上。
如果得到的 DNA 片段大小不合适,改变超声条件,再次进行超声处理。如果得到的 DNA 片段大小合适,继续按步骤 5 所述进行 DMA 末端修补。
喷雾断裂 DNA(请参见图 12-4)
a. 准备如下 DNA 溶液,并置于喷雾杯中。
DNA 样品(来源于步骤 3 ) 5~10ug
10XTM 缓冲液(pH8.0) 200ul
无菌 100% 甘油 1 ml
无菌水 至终体积为 2 ml
b. 将 DNA 样品置于冰浴中,依据经验用最适条件喷雾处理 DNA 样品。
DNA 片段的大小主要决定于氮气的压力。例如,8psi(0.56 kg/cm2) 压力处理黏粒栽体 DNA 主要产生 1000~2500bp 大小的片段。对一个新的靶 DNA 的最适喷雾压力和喷雾时间要依据经验确定。同超声处理-样,冰水浴可以防止 DNA 降解,同时对产生大小均匀的 DNA 片段有重要作用。
C. 将喷雾器置于合适的离心机转头中,用聚苯乙烯泡沫塑料做衬蛰。于 4°C 2000 g(配备微孔板放置装置的离心机用 1000r/min) 快速离心使样品 DNA 溶液沉积于喷雾杯的底部。
d. 将 DNA 样品溶液分成四份,转入 1.5 ml 离心管中,按标准方法乙醇沉淀 DNA, 真空干燥 DNA 沉淀。
e. 用 35ul TE(pH7.6) 溶解每一份 DNA 沉淀,取 1ul 喷雾处理的 DNA 样品和适当分子质量大小的 DNA 分子质量标准,用 0.7% 琼脂糖凝胶电泳分析 DNA 片段大小。电泳时余下的 DNA 样品 4°C 保存。
如果得到的 DMA 片段大小不合适,改变喷雾条件,再次进行喷雾处理。如果得到的 DNA 片段大小合适,继续按步骤 5 所述进行 DNA 末端修补。
DNA 的修补、磷酸化和大小选择
喷雾处理和超声处理产生的 DMA 末端是高度不均一的,包括平末端和残损末端、带有磷酸基团和不带磷酸基团的末端。因为在这些分子中只有一部分能被 DNA 聚合酶修补,流体静力剪切的 DNA 克隆到 M13 噬菌体上的效率通常较低。不过超声处理 5~10ug 靶 DNA, 经过修补和选择大小适当的片段,一般能形成几千个重组克隆。
5. 在打断的 DNA(约 25ul) 中,加入:
10XT4 噬菌体 DNA 聚合酶缓冲液 4.0ul
2.0 mmol/L 含四种 dNTP 溶液 4.0ul
T4 噬菌体 FJNA 聚合酶 5 单位
水 至终体积 40ul
于室温温育 15 min,然后加入约 5 单位 Klenow 片段,继续于室温温育 15 min。
该反应使用两种 DNA 聚合酶修补由于流体静力剪切产生的 DNA 片段的残损末端。T4 噬菌体 DNA 聚合酶补平 3'凹端,并且其外切酶活性可去除突出端。Klenow 片段提供了第二种补平 3'凹端的工具. 有关内容可参见信息栏"大肠杆菌 DNA 聚合酶 Ⅰ Klenow 片段"部分。
6. 用酚: 氯仿抽提纯化 DNA。上层水相转移到一个干净试管中,使 NaCl 浓度达到 0.1mol/L, 加 2 倍体积乙醇,回收沉淀 DNA。用 70% 乙醇洗 DNA 沉淀。
7. 加入 25ul TE(pH7.6) 重新溶解沉淀 DNA。
8. 在微量离心管中混合如下组分:
打断的 DNA 23ul
10x 多核苷酸激酶缓冲液 3ul
20 mmol/L.ATP 3ul
T4 噬菌体多核苷酸激酶 1 单位
T4 噬菌体多核苷酸激酶催化平末端 DNA5'端磷酸化,此步骤不是必需的. 但是一般情況下可以提高 DNA 片段与载体的连接效率。
9. 于 37°C 温育 30 min。
10. 用 0.8% 低熔点琼脂糖凝胶电泳或 5% 中性聚丙烯酰胺凝胶电泳(参见第 5 章)纯化所需大小(0.8~1.5kb)DNA 片段。
为最大限度地减少污染的可能性,应在断裂靶 DNA 和标准参照物之间空出几个泳道,这一点在使用平端 DNA 作为参照物时尤为重要,因其与载体 DNA 的连接效率明显高于剪切的靶 DNA。
11. 用第 5 章描述的方法从凝胶上回收靶 DNA。用 25ulTE(pH7.6) 溶解纯化的 DNA。
12. 取 1ul DNA 用 1% 琼脂糖凝胶电泳分析所纯化 DNA 的完整性和回收率。
与载体 DNA 连接
13. 设立一系列试验连接反应,其中含有 50ng(~0.01pmol) 线性化和去磷酸化载体
DNA 及浓度递增的平端、磷酸化的靶 DNA 片段(见表 12-2)。
14. 连接液通过电转移或转化适当菌株的大肠杆菌感受态细胞(参见第1章的方法23~26)。将细菌铺于含IPTG和X-gal的培养基上,37°C培养过夜。
这一步骤的目的在于找出一个靶DNA片段的浓度,以便最大限度地减小含有人为融合的靶片段的重组体,而后者会使测序更为复杂。因此,当确定大量的连接反应时(步骤16), 必规注意避免使用饱和量的靶DNA。相反,所定出的靶DNA量应为重组克隆的数目与背最相比有中等程度的提高(大约提高至5倍)。
15. 次日计数蓝噬菌斑和白噬菌斑的数目。
采用断裂的平端靶DNA所得的重组体数目大约仅为采用限制性内切核酸酶制备的平端DNA所得数目的十分之一{例如同用Alu I消化的λDNA或ΦX174DNA作为起始连接反应相比)。
16. 用最小量的断裂的平端靶DNA设立一个较大规模的连接反应,以便获得足够的重组克隆完成测序任务。用连接的 DNA转化大肠杆菌,37°C培养过夜。
这一步骤的目的在于确保毎个靶片段至少获得五个重组充隆。图12-5表明了覆盖95% 的双链靶DNA序列所必须测定的克隆的大概数目。
17, 次日收集平板,将转化子保存在适当条件下备用。从一系列单个无色噬菌斑中制备DNA 模板的方法见第 3 章的方法4。
M13重组噬菌体噬菌斑应尽快挑出和扩增(参见第 3 章的方案 2的「噬菌斑的挑取」专栏)。因为噬菌体顆粒可以在顶层琼脂中扩散至较远距离,在37°C生长长时间(>12~16 小时)的噬菌斑或者在 4°C 存放数天的噬菌斑往往已被污染。因此使用从时间较久的噬菌斑制备 DNA 进行测序反应时,背景带的强度和数目都有所增加。
用 96 管盒培养 M13 噬菌体重组克隆
按第3章的方案2 所述方法挑取单一无色噬菌斑,接种在含有 2 ml 细菌培养基的 15 ml 试管中进行培养。然后分别回收备重组病毒颗粒,纯化 DNA(参见第3章的方案 3 和 4)。由于每次只能处理 12 或 24 个克隆,模板纯化过程费时且繁琐。大规模的 DNA 测序需要成千上万的 DNA 模板,但采用有机溶剂抽提和多步离心方法不能快速、经济地制备单链 DNA 模板。取而代之的 M13 噬菌体模板大量制备方法通常包括噬菌体颗粒的纯化或结合自动化技术设备(e.g.,please see Mardis and Roe1989;Smith et al.1990) 用过滤法(Eperon1996)、磁珠法(Alderton et al.1992;Wahlberg et al.1992;Hawkins et al.1994) 或顺磁球法(Fryetal.1992;Wilson1993) 纯化单链 DNA。但是这些设备和操作设备所需的工作人员是一般的学术机构所不具备的。不过 Zollo 和 Chen(1994) 报告了一种用于在 96 孔板中培养 M13 噬菌体克隆,并制备单链 DNA 的有效的可重复的方法,满足了基于荧光的自动化 DNA 测序仪对大量模板的需求。
在鸟枪法测序中,使用一次性平底孔板或一次性试管组盒,以少 M 细菌培养物培养 M13 噬菌体重组子,同时处理 96 个样品是非常有效的。所有单链 DNA 模板制备有关的后续步骤都可以在同一块板中进行,减少了一步又一步转移克隆的工作量,使一个研究人员可在一天内制备 960 个或更多的DNA模板(Pleasesee Zollo and Chen1994)。
18. 对 96 个克隆,每一个克隆都逐一地将大肠杆菌 F'菌株(如 XLl-Blue、XLl-Blue MRF'或 HD5αF') 的单一菌落接种到 100 mlLB 或 2 x YT 培养基,并加至容量为 500 ml 的培养瓶中。于 37°C 以 300r/min 振荡培养 6~8 h。
19. 在培养液中加入 MgS04 至终浓度为 5 mmol/L。
为了保持离心时的平衡和对称,培养的 M13 噬菌体的克隆数为 96 的偶数倍。
Mg2+能提高 M13 噬菌体产量,并降低不同克隆间生长速度差异。
毎组 96 个 M13 噬菌体克隆约需要 80 ml 培养细胞。
20. 用多道移液器转移 0.8 ml 细胞培养液至 96 管盒的各管中。
21. 带上手套,用无菌牙签在每一个 96 管盒的各管中接种单一无色的 M13 噬菌斑。用牙签穿刺噬菌斑的中央,然后投入培养管中。
22. 为避免造成混乱,将牙签保留在培养管中直到所有 96 个培养管都接种完毕。
23. 当最后一个噬菌斑挑取完成,取出所有牙签,封闭培养管盒。培养管盒做好标记,放入 37°C 摇床中,以 250~300r/min 振荡培养 8~12 h。如果需要可重复步骤 20 和 21。
如果培养时间超过 12 h, 制备的单链模板将会被大量的 M13 噬菌体双链复制型 DNA 和/或染色体 DNA 所污染。来源于细菌裂解产生的 DNA 将加大双脱氧测序反应中错配引物的机会。长时间培养也将加大 DNA 克隆片段缺失和重排的机会。因此,适当短时间培养是本方法成功的关键。
M13DNA 的纯化
24. 从培养箱中取出培养管盒,2400 g(带有微量板套筒的离心机转速为 3000~3250r/min) 离心 20 min 沉淀菌体。
M13 噬菌体克隆的主要保存方法是在 50ul 悬液中加人 25ul80% 的甘油,用衡量加样器上下吹打混合溶液,贮存板保存于-80°C。如果需要再制备模板 DNA,这些板作为感染用的噬菌体来源。本方案最后要强调的是安全地保存噬菌体,在本方案中各管间有较大的交叉污染的可能,这对 DNA 测序影响不大,但对于种源的保存是不能接受的。
25. 用多道移液器在新的 96 管盒的各管中加入 120ul 含 20%PEG8000 的 2.5 ml/L NaCl 溶液。
26. 从离心机小心取出离心管,用多道移液器从各管中分别吸取 0.6 ml 上清液加入到已加有 PEG/NaCl 溶液的各管中。
重要:在本步骤中一定不要搅动菌体沉淀,混有菌体将显著降低 DNA 测序质量。
27. 用 96 管盖盖上装有噬菌体悬液和 FEG/NaCl 溶液的试管,确保形成牢固的液封,颠倒试管几次,以混合内容物。试管盒室温放置 30 min, 然后冰浴 30~60 min。
28.2400 g(带有微量板套筒的离心机转速为 3000~3250r/min) 离心 30 min, 收集沉淀噬菌体。取出各排试管,倒置于水槽上空干,在每支试管底部可见少量白色沉淀。将试管放回试管盒。
29. 当所有试管被空干,将试管盒倒置在吸水纸上几分钟空干残存的痕量液体。重新换上新吸水纸,将倒置的试管盒与吸水纸放入离心机中,以 300r/min 离心 3~5 min 去除最后残存的痕量 PEG/NaCl 溶液。
30. 从离心机中取出试管盒,检查试管底部是否仍有噬菌体沉淀。每管加入 20ul TTE 缓冲液。
如果唯菌体生长良好,沉淀呈白色不透明状;如果生长不好,沉淀呈带点蓝色不透明状。如果沉淀呈褐色团块,很可能是因为在步骤 26 取菌体量过多。棕色沉淀制备的模板测序所得结果欠佳。
31. 用 3M 银色胶带密封试管口,在多管旋涡振荡器上剧烈振荡试管盒 15~30 min。
32. 快速离心试管盒使液体沉于管底,去掉每一个 96 孔管盒底座,将试管置于 80°C 水浴中保温 10 min。
该步骤是为了裂解 M13 噬菌体颗粒。去掉管盒的底座可以确保所有的试管在 80°C 保温达到 10 min。
33. 从水浴中取出试管并冷却至室温。重新安上试管盒底座,快速离心试管盒使液体沉于管底。
34. 用多道移液器将 70ul 无菌水加到 96 孔板的各孔中,将步骤 33 获得的噬菌体裂解液转移到 96 孔板中,用移液器吹打混匀两种溶液。用一条 3M 银胶带密封 96 孔板,如果在 24~48 h 内进行测序也可以用 96 孔板盖板盖住。标记好各块板,贮存于-20°C。
每进行一次培养可以提取到 2.5~5ulM13 噬菌体单链 DNA。
35. 随机从一些孔中取 DNA 样品(5ul), 用 1% 琼脂糖凝较电泳检査 DNA 量。
如果一切正常,各管中的 DNA 产虽差别很小。DNA 循环测序反应一般需制备的 DNA 量为 2ul~7.5ul
(参见方案 6)。



相关文章

