
cDNA 5’末端的快速扩增(5’-RACE)
原理在分子克隆中没有比分离缺乏相应的 mRNA 5‘ 末端的序列特征结构的 cDNA 克隆更易失败的了。通常存在于 cDNA 基因文库中的部分克隆,由于
原理
在分子克隆中没有比分离缺乏相应的 mRNA 5' 末端的序列特征结构的 cDNA 克隆更易失败的了。通常存在于 cDNA 基因文库中的部分克隆,由于反转录酶未能沿全长 mRNA 模板延伸完全,合成的 cDNA 第一条链往往 5' 末端不完整。
材料与仪器
反转录酶 末端脱氧核苷酸转移酶 末端转移酶缓冲液 热稳定 DNA 聚合酶
扩增缓冲液 氯仿 dATP dNTP 贮存液 胎盘 RNase 抑制剂 TE 反转录酶缓冲液
琼脂糖凝胶或聚丙烯酰胺凝胶 制备型离心机 SorvallSS - 34 转头
扩增缓冲液 氯仿 dATP dNTP 贮存液 胎盘 RNase 抑制剂 TE 反转录酶缓冲液
琼脂糖凝胶或聚丙烯酰胺凝胶 制备型离心机 SorvallSS - 34 转头
步骤
一、材料
1. 缓冲液与溶液
10X 扩增缓冲液
氯仿
dATP ( 1 mmol/L,二钠盐)
4 种 dNTP 贮存液(20 mmol/L, pH 8.0)
胎盘 RNase 抑制剂(20 单位/μl)
TE ( pH 7.6)
2. 酶与缓冲液
5X 反转录酶缓冲液
反转录酶(RNA 依赖的 DNA 聚合酶)
末端脱氧核苷酸转移酶(末端转移酶)
5X末端转移酶缓冲液
热稳定 DNA 聚合酶
3. 凝胶
琼脂糖凝胶或聚丙烯酰胺凝胶
4. 核苷酸与寡核苷酸
接头引物(10 μmol/L,5'GACTCGAGTCGACATCG3')溶于水中(10 pmol/μl)
(接头引物可以与基因特异性正向引物联合在一起用于扩增特异性靶 cDNA。第一步 PCR 扩增后,PCR 目的产物可能仅占总 DNA 产物的 1%,也可能多至占总 DNA 产物的 100%。如果必要,可用第一轮 PCR 产物作模板进行第二轮嵌套式 PCR,改善目的产物的产率,该轮 PCR 的引物为接头引物与另一个基因特异性正向引物。在第二轮嵌套式 PCR 扩增后,几乎所有的扩增产物在 EB 染色下呈现了靶 mRNA 的 5' 区域序列相一致的条带。
RT-PCR 的引物用自动 DNA 合成仪合成,用于标准 RT-PCR 的引物一般不需要进一步纯化。然而,如果寡核苷酸引物经商品化的树脂进行柱层析纯化(如 NENLife- Science 公司产品 NENSORB) 或者经变性的聚丙烯酰胺凝胶电泳纯化,纯化后的引物用于扩增低丰度的 mRNA 时常常会更有效。)
(dT)17-接头引物(10 μmol/L,5'GACTCGAGTCGACATCGA(T)173')溶于水中(10 pmol/μl)
基因特异性反义寡核苷酸引物(10 μmol/L) 溶于水(10 pmol/μl)。
(基因特异性反义寡核苷酸引物应该与靶 mRNA 的已知序列互补,其长度在 20~ 30 bp,内含的 4 种碱基数量基本相等,G 与 C 碱基的平衡分布以及引物具有较低的自发形成稳定二级结构的倾向。基因特异性引物 1 用于步骤 2, 用反转录酶引导基因特异性第一链 cDNA 的合成。基因特异性引物 2 是与靶 mRNA 的 5'序列互补的,用于 5'-RACE 反应的扩增阶段。基因特异性引物 2 也总是被插入适当的限制性内切核酸酶酶切位点,这样便于扩增 cDNA 的进一步克隆。)
随机六核苷酸溶于 TE ( 1 mg/ml,pH 8.0)
总 RNA(100 μg/ml)或 poly (A)+ RNA(10 μg/ml)溶于水中
(总 RNA一般用离液剂(chaotropic agents) 从细胞中抽提的,选择总 RNA 作模板一般适用于从中等程度到高丰度 mRNA 中通过 RT-PCR 克隆靶基因。对于低丰度靶 mRNA 的样品最好选择 poly (A) + RNA 作模板进行 RT-PCR。运用作为 RT-PCR 模板的 RNA 也可以从如下的少量培养细胞中制备。)
5. 离心机与转头
制备型离心机
SorvallSS - 34 转头
6. 特殊设备
自动微量移液器的屏蔽型枪头
浓缩器(Centricon-100,Amicon 产品)
微量离心管(0.5 ml,薄壁扩增反应专用离心管)或微量滴定板
正向排液式移液器
PCR 仪
旋转真空冷冻干燥机(可选择)
水浴箱预设在 75℃ 和 80℃
二、方法
反转录
在实验开始时,首先要确定 5'-RACE 是否成功。如果反转录步骤是有效的,那么分离到含有靶 mRNA 的 5' 末端序列的克隆的几率是较高的。另一方面,如果反转录步骤是无效的,那么本方案的后续步骤是无法补偿的。因此,对于反转录反应条件是值得花时间优化的,如最佳的引物与模板比例,改变反应体系 Mg2+ 浓度,寻找最适的 Mg2+ 浓度。
1. 转移 1 pg 至 100 ng 的 poly (A) + RNA 或 1 μg 总 RNA 到一只新的离心管。用水调整体积至将 RNA 样品在 75℃ 变性 5 min, 将离心管插入冰中冷却。
2. 向这种含有已变性的 RNA 样品的离心管内依次加入如下试剂:
5X 反转录酶缓冲液 4 μl
20 mmol/L 4 种 dNTP 混合液(pH 8.0) 1 μl
10 μmol/L 基因特异性反义引物 1 4 μl
约 20 单位/μl 胎盘 RNase 抑制剂 1 μl
水补足至 20 μl
反应管温育在反应 60 min。设置 3 支阴性对照管。在一支对照管中加入合成第一链 cDNA 反应所需的所有试剂,但不加模板 RNA;第二支对照管加入除了反转录酶外的所有试剂;第三支对照管中加入除引物外的所有试剂。这些对照管进行所有的后续步骤。设置这些对照管能保证 cDNA 产物不是由于基因组 DNA 与寡核苷酸片段的污染或者由 RNA 模板分子的自身引导所致。
反转录产物的纯化
反转录反应完成后,多余的 dNTP 和引物必须从反应液中去除。此外,dNTP 在反应液中的存在,一方面可能由末端转移酶在 cDNA 的 3' 末端加尾,另一方面还可能危及第一链 cDNA 尾部序列作为引物结合位点的能力。多余的引物的存在也会造成不利的影响,因为引物的 3' 末端会与 cDNA 的加尾反应相竞争。
3. 去除过剩的寡核苷酸或随机六核苷酸引物可用水将反应液终体积稀释至 2 ml, 然后用 Centicon-100 微量浓缩器(Frohman 1993; for a description of this procedure, please see protocol 3 )。离心转速为 500~1100 g ( 2000~3000 r/min, 用 Sorvall SS34 转头), 温度在 4℃ 和 25℃ 之间离心 20 min。重复用水稀释步骤和离心步骤。转移潴留的液体到一支新的 0.5 ml 离心管,用旋转真空抽干机使体积浓缩至 10 μl。
4. 加入 10 体积的如下试剂至反转录 cDNA 样品中:
5X 末端转移酶缓冲液 4 μl
1 mmol/L dATP 4 μl
末端转移酶 10~25 单位
反应管孵育在 37℃ 水浴中反应 15 min。
5. 在 80℃ 放置 3 min 使末端转移酶灭活。使带有 dA 尾的 cDNA 样品用 TE pH 7.6 稀释至终体积 1 ml。
扩增
6. 按以下次序,将各成分加入在一只 0.5 ml 灭菌离心管、扩增管或灭菌微量滴定板的孔内,建立 PCR 系列反应管:
已稀释 cDNA 0~20 μl
10X 扩增缓冲液 5 μl
20 mmol/L 4 种 dNTP 混合液 5 μl
10 μmol/L (dT)17-接头引物(16 pmol) 1.6 μl
10 μmol/L 接头引物(32 pmol) 3.2 μl
10 μmol/L 基因特异性引物 2 ( 32 pmol ) 3.2 μl
1~2 单位热稳定 DNA 聚合酶 1 μl
H2O 补足至 50 μl
如果实验必要,可建立系列的扩增试验管用于筛选 cDNA 加尾模板产生最大量的扩增 5' 末端。
7. 如果 PCR 仪没有配置加热盖,在反应混合液的上层应加一滴矿物油(约 50 μl),防止样品在 PCR 反应多个加热与冷却的循环过程中蒸发。如果应用热启动 PCR 程序,在反应混合液上层加一滴石蜡油。放置离心管或微量滴定板在 PCR 仪上。
8. 按以下方法进行 PCR 扩增。典型的程序有变性、复性和聚合(延伸反应);相应的循环条件与温度列表如下:
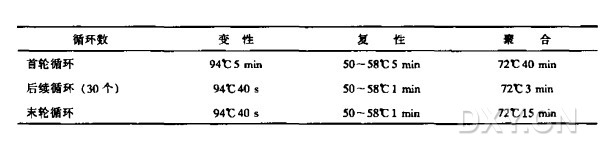
9. 抽取每种扩增样品 5~10 μl,用琼脂凝胶电泳或聚丙烯酰胺凝胶电泳来分析扩增结果,用 DNA marker 来判断扩增片段的大小。凝胶一般用 EB ( 溴化乙锭)或 SYBR 金颗粒染色后来观察扩增的量与片段大小。
10. 若用矿物油覆盖在微量离心管内样品液体的上层(步骤 7),反应结束后可用 150 μl 氯仿抽提去除。
11. 从扩增的 DNA 产物中分离掉残余的热稳定 DNA 聚合酶和 dNTP。

1. 缓冲液与溶液
10X 扩增缓冲液
氯仿
dATP ( 1 mmol/L,二钠盐)
4 种 dNTP 贮存液(20 mmol/L, pH 8.0)
胎盘 RNase 抑制剂(20 单位/μl)
TE ( pH 7.6)
2. 酶与缓冲液
5X 反转录酶缓冲液
反转录酶(RNA 依赖的 DNA 聚合酶)
末端脱氧核苷酸转移酶(末端转移酶)
5X末端转移酶缓冲液
热稳定 DNA 聚合酶
3. 凝胶
琼脂糖凝胶或聚丙烯酰胺凝胶
4. 核苷酸与寡核苷酸
接头引物(10 μmol/L,5'GACTCGAGTCGACATCG3')溶于水中(10 pmol/μl)
(接头引物可以与基因特异性正向引物联合在一起用于扩增特异性靶 cDNA。第一步 PCR 扩增后,PCR 目的产物可能仅占总 DNA 产物的 1%,也可能多至占总 DNA 产物的 100%。如果必要,可用第一轮 PCR 产物作模板进行第二轮嵌套式 PCR,改善目的产物的产率,该轮 PCR 的引物为接头引物与另一个基因特异性正向引物。在第二轮嵌套式 PCR 扩增后,几乎所有的扩增产物在 EB 染色下呈现了靶 mRNA 的 5' 区域序列相一致的条带。
RT-PCR 的引物用自动 DNA 合成仪合成,用于标准 RT-PCR 的引物一般不需要进一步纯化。然而,如果寡核苷酸引物经商品化的树脂进行柱层析纯化(如 NENLife- Science 公司产品 NENSORB) 或者经变性的聚丙烯酰胺凝胶电泳纯化,纯化后的引物用于扩增低丰度的 mRNA 时常常会更有效。)
(dT)17-接头引物(10 μmol/L,5'GACTCGAGTCGACATCGA(T)173')溶于水中(10 pmol/μl)
基因特异性反义寡核苷酸引物(10 μmol/L) 溶于水(10 pmol/μl)。
(基因特异性反义寡核苷酸引物应该与靶 mRNA 的已知序列互补,其长度在 20~ 30 bp,内含的 4 种碱基数量基本相等,G 与 C 碱基的平衡分布以及引物具有较低的自发形成稳定二级结构的倾向。基因特异性引物 1 用于步骤 2, 用反转录酶引导基因特异性第一链 cDNA 的合成。基因特异性引物 2 是与靶 mRNA 的 5'序列互补的,用于 5'-RACE 反应的扩增阶段。基因特异性引物 2 也总是被插入适当的限制性内切核酸酶酶切位点,这样便于扩增 cDNA 的进一步克隆。)
随机六核苷酸溶于 TE ( 1 mg/ml,pH 8.0)
总 RNA(100 μg/ml)或 poly (A)+ RNA(10 μg/ml)溶于水中
(总 RNA一般用离液剂(chaotropic agents) 从细胞中抽提的,选择总 RNA 作模板一般适用于从中等程度到高丰度 mRNA 中通过 RT-PCR 克隆靶基因。对于低丰度靶 mRNA 的样品最好选择 poly (A) + RNA 作模板进行 RT-PCR。运用作为 RT-PCR 模板的 RNA 也可以从如下的少量培养细胞中制备。)
5. 离心机与转头
制备型离心机
SorvallSS - 34 转头
6. 特殊设备
自动微量移液器的屏蔽型枪头
浓缩器(Centricon-100,Amicon 产品)
微量离心管(0.5 ml,薄壁扩增反应专用离心管)或微量滴定板
正向排液式移液器
PCR 仪
旋转真空冷冻干燥机(可选择)
水浴箱预设在 75℃ 和 80℃
二、方法
反转录
在实验开始时,首先要确定 5'-RACE 是否成功。如果反转录步骤是有效的,那么分离到含有靶 mRNA 的 5' 末端序列的克隆的几率是较高的。另一方面,如果反转录步骤是无效的,那么本方案的后续步骤是无法补偿的。因此,对于反转录反应条件是值得花时间优化的,如最佳的引物与模板比例,改变反应体系 Mg2+ 浓度,寻找最适的 Mg2+ 浓度。
1. 转移 1 pg 至 100 ng 的 poly (A) + RNA 或 1 μg 总 RNA 到一只新的离心管。用水调整体积至将 RNA 样品在 75℃ 变性 5 min, 将离心管插入冰中冷却。
2. 向这种含有已变性的 RNA 样品的离心管内依次加入如下试剂:
5X 反转录酶缓冲液 4 μl
20 mmol/L 4 种 dNTP 混合液(pH 8.0) 1 μl
10 μmol/L 基因特异性反义引物 1 4 μl
约 20 单位/μl 胎盘 RNase 抑制剂 1 μl
水补足至 20 μl
反应管温育在反应 60 min。设置 3 支阴性对照管。在一支对照管中加入合成第一链 cDNA 反应所需的所有试剂,但不加模板 RNA;第二支对照管加入除了反转录酶外的所有试剂;第三支对照管中加入除引物外的所有试剂。这些对照管进行所有的后续步骤。设置这些对照管能保证 cDNA 产物不是由于基因组 DNA 与寡核苷酸片段的污染或者由 RNA 模板分子的自身引导所致。
反转录产物的纯化
反转录反应完成后,多余的 dNTP 和引物必须从反应液中去除。此外,dNTP 在反应液中的存在,一方面可能由末端转移酶在 cDNA 的 3' 末端加尾,另一方面还可能危及第一链 cDNA 尾部序列作为引物结合位点的能力。多余的引物的存在也会造成不利的影响,因为引物的 3' 末端会与 cDNA 的加尾反应相竞争。
3. 去除过剩的寡核苷酸或随机六核苷酸引物可用水将反应液终体积稀释至 2 ml, 然后用 Centicon-100 微量浓缩器(Frohman 1993; for a description of this procedure, please see protocol 3 )。离心转速为 500~1100 g ( 2000~3000 r/min, 用 Sorvall SS34 转头), 温度在 4℃ 和 25℃ 之间离心 20 min。重复用水稀释步骤和离心步骤。转移潴留的液体到一支新的 0.5 ml 离心管,用旋转真空抽干机使体积浓缩至 10 μl。
4. 加入 10 体积的如下试剂至反转录 cDNA 样品中:
5X 末端转移酶缓冲液 4 μl
1 mmol/L dATP 4 μl
末端转移酶 10~25 单位
反应管孵育在 37℃ 水浴中反应 15 min。
5. 在 80℃ 放置 3 min 使末端转移酶灭活。使带有 dA 尾的 cDNA 样品用 TE pH 7.6 稀释至终体积 1 ml。
扩增
6. 按以下次序,将各成分加入在一只 0.5 ml 灭菌离心管、扩增管或灭菌微量滴定板的孔内,建立 PCR 系列反应管:
已稀释 cDNA 0~20 μl
10X 扩增缓冲液 5 μl
20 mmol/L 4 种 dNTP 混合液 5 μl
10 μmol/L (dT)17-接头引物(16 pmol) 1.6 μl
10 μmol/L 接头引物(32 pmol) 3.2 μl
10 μmol/L 基因特异性引物 2 ( 32 pmol ) 3.2 μl
1~2 单位热稳定 DNA 聚合酶 1 μl
H2O 补足至 50 μl
如果实验必要,可建立系列的扩增试验管用于筛选 cDNA 加尾模板产生最大量的扩增 5' 末端。
7. 如果 PCR 仪没有配置加热盖,在反应混合液的上层应加一滴矿物油(约 50 μl),防止样品在 PCR 反应多个加热与冷却的循环过程中蒸发。如果应用热启动 PCR 程序,在反应混合液上层加一滴石蜡油。放置离心管或微量滴定板在 PCR 仪上。
8. 按以下方法进行 PCR 扩增。典型的程序有变性、复性和聚合(延伸反应);相应的循环条件与温度列表如下:
9. 抽取每种扩增样品 5~10 μl,用琼脂凝胶电泳或聚丙烯酰胺凝胶电泳来分析扩增结果,用 DNA marker 来判断扩增片段的大小。凝胶一般用 EB ( 溴化乙锭)或 SYBR 金颗粒染色后来观察扩增的量与片段大小。
10. 若用矿物油覆盖在微量离心管内样品液体的上层(步骤 7),反应结束后可用 150 μl 氯仿抽提去除。
11. 从扩增的 DNA 产物中分离掉残余的热稳定 DNA 聚合酶和 dNTP。



相关文章

