
降落 PCR基本方案
原理
基于PCR原理三步骤而设置变性-退火-延伸三个温度点。在标准反应中采用三温度点法,双链DNA在90~95℃变性,再迅速冷却至40 ~60℃,引物退火并结合到靶序列上,然后快速升温至70~75℃,在Taq DNA 聚合酶的作用下,使引物链沿模板延伸。对于较短靶基因(长度为100~300bp时)可采用二温度点法, 除变性温度外、退火与延伸温度可合二为一,一般采用94℃变性,65℃左右退火与延伸(此温度Taq DNA酶仍有较高的催化活性)。
①变性温度与时间:变性温度低,解链不完全是导致PCR失败的最主要原因。一般情况下,93℃~94℃lmin足以使模板DNA变性,若低于93℃则需延长时间,但温度不能过高,因为高温环境对酶的活性有影响。此步若不能使靶基因模板或PCR产物完全变性,就会导致PCR失败。
②退火(复性)温度与时间:退火温度是影响PCR特异性的较重要因素。变性后温度快速冷却至40℃~60℃,可使引物和模板发生结合。由于模板DNA 比引物复杂得多,引物和模板之间的碰撞结合机会远远高于模板互补链之间的碰撞。退火温度与时间,取决于引物的长度、碱基组成及其浓度,还有靶基序列的长度。对于20个核苷酸,G+C含量约50%的引物,55℃为选择最适退火温度的起点较为理想。引物的复性温度可通过以下公式帮助选择合适的温度:
Tm值(解链温度)=4(G+C)+2(A+T)
复性温度=Tm值-(5~10℃)
在Tm值允许范围内, 选择较高的复性温度可大大减少引物和模板间的非特异性结合, 提高PCR反应的特异性。复性时间一般为30~60sec,足以使引物与模板之间完全结合。
③延伸温度与时间:Taq DNA聚合酶的生物学活性:
70~80℃ 150核苷酸/S/酶分子
70℃ 60核苷酸/S/酶分子
55℃ 24核苷酸/S/酶分子
高于90℃时, DNA合成几乎不能进行。
PCR反应的延伸温度一般选择在70~75℃之间,常用温度为72℃,过高的延伸温度不利于引物和模板的结合。PCR延伸反应的时间,可根据待扩增片段的长度而定,一般1Kb以内的DNA片段,延伸时间1min是足够 的。3~4kb的靶序列需3~4min;扩增10Kb需延伸至15min。延伸进间过长会导致非特异性扩增带的出现。对低浓度模板的扩增,延伸时间要稍长些。
循环次数 循环次数决定PCR扩增程度。PCR循环次数主要取决于模板DNA的浓度。一般的循环次数选在30~40次之间,循环次数越多,非特异性产物的量亦随之增多。
材料与仪器
引物 去离子水 DNA聚合酶 缓冲液 dNTP MgSO4(可选)和DMSO(可选)
PCR
步骤
2. 人CD137胞膜外区基因的PCR扩增体系:CD137-pCDNA3质粒4 ul,P1,P2各0.5 umol/L,MgCl2 2 mmol/L,dNTP 0.4 mmol/L,Taq 5 U。
3. hIgG1Fc片段的PCR扩增体系:含人hIgG1Fc的PGEM-T质粒4 ul,P3、P4各0.4 umol/L,MgCl2 2 mmol/L,dNTP 0.4 mmol/L,Taq 5 U。
4. HBVDNA多聚酶基因的PCR扩增体系:
(1)第一轮:HBVDNA模板4 ul,P5、P6各0.25 umol/L,MgCl2 1.5 mmol/L,dNTP 0.2 mmol/L,Taq 0.8 U.
(2)第二轮:取5倍稀释的第一轮PCR产物4 ul为模板,P7、P8各0.25 umol/L,MgCl2 1.5 mmol/L,dNTP 0.2 mmol/L,Taq 0.8 U。
5. 分别在各灭菌PCR管加入上述各反应成分,100℃煮沸5 min,立即冰浴5 min,加入TaqDNA聚合酶,混匀,离心,PCR程序如下:
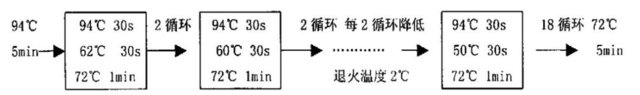
注意事项
常见问题
1、若无条带:
(1)反应体系错误。重新配置反应体系确保各种试剂加入的量没有问题。
(2)PCR程序出错。检测PCR仪程序是否正确。
(3)DNA胶问题。DNA凝胶电泳时加入阳性对照。
(4)退火温度不合适。以2度为梯度设计梯度PCR反应优化退火温度。
(5)DNA模板量太少。增加DNA模板量。
(6)引物错误。利用BLAST检查引物特异性或重新设计引物。
(7)DNA模板中存在抑制剂。确保DNA模板干净
(8)模板有复杂结构。加入DMSO,BSA或者甜菜碱。可尝试递减PCR。
2、PCR产物量过少:
(1)延伸时间太短。以1kb/分钟的原则设置延伸时间。
(2)变性时间过长。变性时间过长会导致DNA聚合酶失活。


相关文章
- RT-PCR、Western blot和ELISA三者的区别
- PCR反应五要素
- 原位聚合酶链式反应(in situ PCR)和原位反转录聚合酶链式反应(in situ RT-PCR)操作规程
- A sensitive quantification of HHV-6B by real-time PCR
- PCR标准反应体系
- Validation of RNAi by Real Time PCR
- Site-Directed Mutagenesis and Gene Fusion by Megaprimer PCR
- 逆转录-聚合酶链反应 (Reverse Transcription-Polymerase Chain Reaction,RT-PCR
- 锚定PCR
- Real-Time PCR Fluorescent Chemistries

