
从全血中纯化基因组 DNA 实验方法
从全血中纯化基因组 DNA 实验方法
材料与仪器
乙醇 异丙醇 RNA 酶 A 全血
琼脂糖凝胶电泳 离心管 Wizard 基因组 DNA 纯化试剂盒
琼脂糖凝胶电泳 离心管 Wizard 基因组 DNA 纯化试剂盒
步骤
一、材料
1. 缓冲液、溶液和试剂
乙醇,70%,室温
异丙醇, 室温
RNA 酶 A
将 RNA 酶 A 溶解于 DNA 再水合液中至终浓度为 4 mg/ml, 煮沸 10min 以去除 DNA 酶的污染。分装后保存于-20°C。
2. 专用设备
琼脂糖凝胶电泳设备
50 ml 的消毒离心管
3. 其他
Wizard 基因组 DNA 纯化试剂盒(Promega; 包括细胞裂解液、核裂解液、蛋白质沉淀液和 DNA 再水合液)
分子量标记,用于琼脂糖凝肢电泳
4. 细胞和组织
全血 (10 ml)
二、方法
1. 血红细胞的裂解
(1) 在 50 ml 已消毒的离心管中加入 30 ml 细胞裂解液
(2) 轻轻摇动血液样品管至样品完全混合;然后将血样 (10 ml) 转移到已加有细胞裂解液的管中,将管颠倒 5 或 6次使之混合,
(3) 将混合物在室温下温育 l0min(期间颠倒混合 3 次)以裂解血红细胞,室温下以 2000 g 离心 l0min。
(4) 在不破坏白色可见沉降物的前提下尽量移除上液,大约会有 1.4 ml 的残液。如果血样已经凝固再加入
30 ml 细胞裂解液,颠倒 5 或 6 次混合。重复 (3) 和 (4) 直到沉降物几乎完全变白。从凝固样品中提取 DNA 可能会有一些损失。
在步骤 (4) 中,可能会看到白细胞中也漫有一些血红细胞或细胞碎片。如果沉降物中似乎只含有血红细胞,那么弃上清液后在细胞沉降物中再加入 30 ml 细胞裂解液,然后重复 (3) 和 (4)。
(5) 剧烈振荡试管直到白细胞重悬于其中(10~15s)。
2. 核的裂解和蛋白质的沉淀
(1) 向含有重悬细胞的试管中加入 l0 ml 核裂解液,抽吸溶液 5 或 6 次使白细胞裂解,这样溶液将会变得非常黏滞。混合后如果有成块细胞出现,那么将溶液放置于37°C 进行温育直至细胞块完全破坏。若 lh 后仍然能够看到细胞块,则再加 3 ml 核裂解液,反复温育处理。
(2) 可选择的步骤:向核裂解产物中加入 RNA 酶 A20ug/ml, 颠倒试管 2?5 次进行混合,混合物在 37°C 温育 15 min, 然后冷却至室溫。
(3) 向核裂解产物中加入 3.3 ml 蛋白质沉淀液,剧烈荡 10?20s 后可能会看到小的蛋白质块。
(4) 室温下 2000 g 离心 lOmin 后将看到一个深褐色的白质块。
3.DNA 的沉淀和再水合
(1) 室温下,将上清液转移到一个装有 10 ml 异丙醇的50 ml 干净试管中。
(2) 轻轻颠倒混合溶液直至白色线状 DNA 变形可见的块状 DNA。
(3) 室温下以 2000 g 离心 1 min 后将会有小块的白色 DNA 沉降出现
(4) 室温下慢慢倒出上清液,加入 l0 ml70% 乙酵,轻柔地颠倒试管数次以洗涤 DNA 沉淀和试管壁,然后按照 (3) 进行离心。
(5) 小心吸出乙醇。此时的 DNA 沉降非常松散,必须小心避免将沉降物吸到移液管中
将试管颠倒后放置在干净的吸水纸上,风干沉降物 10~15 min。
(6) 加入 800ulDNA 再水合液,65°C 温育 lh 使 DNA 再水合,期间定时轻敲试管以使溶液混合。再水合 DNA 的另一个方法是在室温或 4°C 将溶液温育过夜。
(7) 在 2~8°C 储存 DNA。

可以通过测定 DNA 在 260mn 下的吸光度或者通过琼脂糖凝胶电泳来测定纯化样品相对于标准样的量等方法来对 DNA 进行定量。如果需要精确的浓度,推荐使用后一种方法而不是由分光光度计来确定,因为吸光度的读数可因样品中低分子量紫外线吸收物质的存在而偏高(Glasel1995)。通常 10 ml 全血的 DNA 产量为 250~500ul(表 9-1)。
1. 缓冲液、溶液和试剂
乙醇,70%,室温
异丙醇, 室温
RNA 酶 A
将 RNA 酶 A 溶解于 DNA 再水合液中至终浓度为 4 mg/ml, 煮沸 10min 以去除 DNA 酶的污染。分装后保存于-20°C。
2. 专用设备
琼脂糖凝胶电泳设备
50 ml 的消毒离心管
3. 其他
Wizard 基因组 DNA 纯化试剂盒(Promega; 包括细胞裂解液、核裂解液、蛋白质沉淀液和 DNA 再水合液)
分子量标记,用于琼脂糖凝肢电泳
4. 细胞和组织
全血 (10 ml)
二、方法
1. 血红细胞的裂解
(1) 在 50 ml 已消毒的离心管中加入 30 ml 细胞裂解液
(2) 轻轻摇动血液样品管至样品完全混合;然后将血样 (10 ml) 转移到已加有细胞裂解液的管中,将管颠倒 5 或 6次使之混合,
(3) 将混合物在室温下温育 l0min(期间颠倒混合 3 次)以裂解血红细胞,室温下以 2000 g 离心 l0min。
(4) 在不破坏白色可见沉降物的前提下尽量移除上液,大约会有 1.4 ml 的残液。如果血样已经凝固再加入
30 ml 细胞裂解液,颠倒 5 或 6 次混合。重复 (3) 和 (4) 直到沉降物几乎完全变白。从凝固样品中提取 DNA 可能会有一些损失。
在步骤 (4) 中,可能会看到白细胞中也漫有一些血红细胞或细胞碎片。如果沉降物中似乎只含有血红细胞,那么弃上清液后在细胞沉降物中再加入 30 ml 细胞裂解液,然后重复 (3) 和 (4)。
(5) 剧烈振荡试管直到白细胞重悬于其中(10~15s)。
2. 核的裂解和蛋白质的沉淀
(1) 向含有重悬细胞的试管中加入 l0 ml 核裂解液,抽吸溶液 5 或 6 次使白细胞裂解,这样溶液将会变得非常黏滞。混合后如果有成块细胞出现,那么将溶液放置于37°C 进行温育直至细胞块完全破坏。若 lh 后仍然能够看到细胞块,则再加 3 ml 核裂解液,反复温育处理。
(2) 可选择的步骤:向核裂解产物中加入 RNA 酶 A20ug/ml, 颠倒试管 2?5 次进行混合,混合物在 37°C 温育 15 min, 然后冷却至室溫。
(3) 向核裂解产物中加入 3.3 ml 蛋白质沉淀液,剧烈荡 10?20s 后可能会看到小的蛋白质块。
(4) 室温下 2000 g 离心 lOmin 后将看到一个深褐色的白质块。
3.DNA 的沉淀和再水合
(1) 室温下,将上清液转移到一个装有 10 ml 异丙醇的50 ml 干净试管中。
(2) 轻轻颠倒混合溶液直至白色线状 DNA 变形可见的块状 DNA。
(3) 室温下以 2000 g 离心 1 min 后将会有小块的白色 DNA 沉降出现
(4) 室温下慢慢倒出上清液,加入 l0 ml70% 乙酵,轻柔地颠倒试管数次以洗涤 DNA 沉淀和试管壁,然后按照 (3) 进行离心。
(5) 小心吸出乙醇。此时的 DNA 沉降非常松散,必须小心避免将沉降物吸到移液管中
将试管颠倒后放置在干净的吸水纸上,风干沉降物 10~15 min。
(6) 加入 800ulDNA 再水合液,65°C 温育 lh 使 DNA 再水合,期间定时轻敲试管以使溶液混合。再水合 DNA 的另一个方法是在室温或 4°C 将溶液温育过夜。
(7) 在 2~8°C 储存 DNA。

4. 分离 DNA 的定性和定量
可以通过测定 DNA 在 260mn 下的吸光度或者通过琼脂糖凝胶电泳来测定纯化样品相对于标准样的量等方法来对 DNA 进行定量。如果需要精确的浓度,推荐使用后一种方法而不是由分光光度计来确定,因为吸光度的读数可因样品中低分子量紫外线吸收物质的存在而偏高(Glasel1995)。通常 10 ml 全血的 DNA 产量为 250~500ul(表 9-1)。

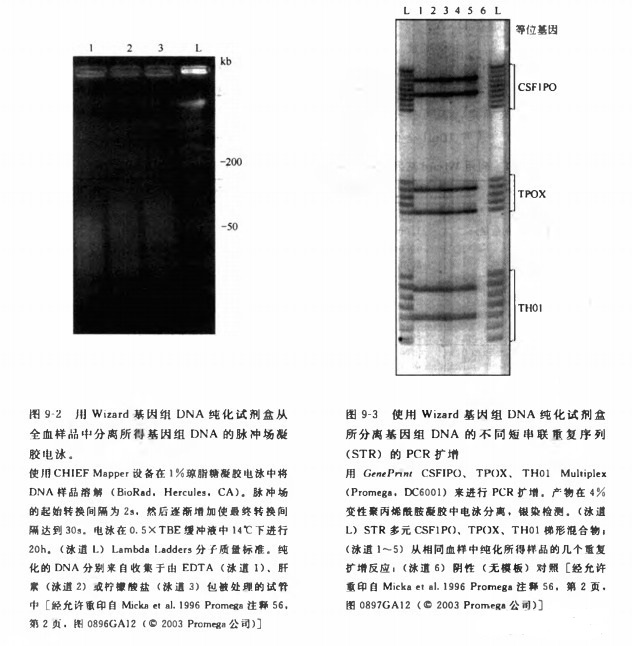
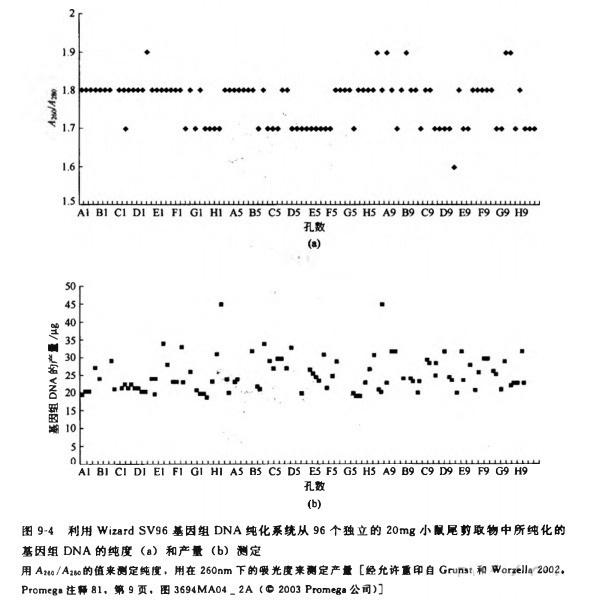
用该方法分离的 DNA 质量可以通过测定扩增的长度和 PCR 扩增的能力来估测(Saikiet al.1985) 脉冲场凝胶电泳结果显示由该方法分离的 DNA 片段绝大部分在 50?200kb(图9-2)。所分离 DNA 与 PCR 扩增的兼容性可以通过对 3 个人多态性位点的成功扩增来证明。对来自 5 个不同个体的样品(25ug)都进行了高效扩增,每个位点都产生相同类型的平行带(图 9-3)。


相关文章
- RT-PCR、Western blot和ELISA三者的区别
- PCR反应五要素
- 原位聚合酶链式反应(in situ PCR)和原位反转录聚合酶链式反应(in situ RT-PCR)操作规程
- A sensitive quantification of HHV-6B by real-time PCR
- PCR标准反应体系
- Validation of RNAi by Real Time PCR
- Site-Directed Mutagenesis and Gene Fusion by Megaprimer PCR
- 逆转录-聚合酶链反应 (Reverse Transcription-Polymerase Chain Reaction,RT-PCR
- 锚定PCR
- Real-Time PCR Fluorescent Chemistries

