
粘性末端克隆
原理
限制性内切酶能够识别并酶切双链 DNA 分子中的特定核苷酸序列,使目的 DNA 片段和载体产生不平齐的粘性末端,然后在 DNA 连接酶的作用下,将酶切后的目的 DNA 片段和载体进行连接,构成重组载体。
用途
构建质粒。
材料与仪器
目的 DNA 或者 cDNA、引物(带有酶切位点和保护碱基等)
PCR 试剂(天根 KT211)、载体质粒、限制性内切酶(Thermo Scientific)
琼脂糖、DNA 琼脂糖凝胶回收试剂盒、T4 DNA 连接酶(Takara 2011A)
大肠杆菌、LB 平板和 LB 肉汤培养基、氨苄或者卡那抗生素、质粒抽提试剂盒
步骤
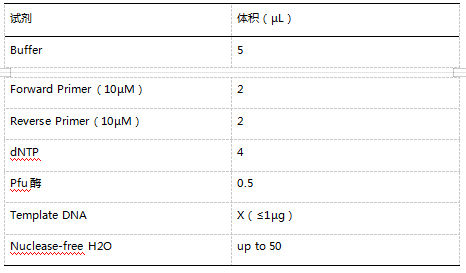
(1)PCR:按照 PCR 试剂说明书(天根 KT211),加入目的 DNA 或者 cDNA、引物、PCR 试剂、ddH2O 等,按照程序进行 PCR 扩增。参考体系如下:
程序如下:

扩增完成后,将 PCR 产物按 120 V 30 min 的条件进行琼脂糖凝胶电泳,一般情况下琼脂糖凝胶的浓度为 0.8%~1.0%。在核酸凝胶成像仪中观察目的条带,将目的凝胶切割下来,按照 DNA 琼脂糖凝胶回收试剂盒进行回收,获得目的 DNA 片段。
(2)酶切:按照限制性内切酶说明书(Thermo Scientific),将目的 DNA 片段和载体质粒进行酶切,通常 1~2 μg 质粒在 37 ℃ 酶切 2.5~3 h 足够,将酶切后的目的 DNA 片段和载体进行琼脂糖凝胶电泳和回收。
(3)连接:按照 T4 DNA 连接酶(Takara 2011A)说明书进行连接,目的 DNA 片段与载体的摩尔比建议为 3:1 - 6:1,总质量控制在 100~200 ng,4 ℃ 连接过夜或者 16 ℃ 2~3 h。

(4)质粒转化:从 -80 ℃ 冰箱取出感受态细胞(大肠杆菌,如 DH5α)置于冰上融化,加入连接产物轻轻混匀后放置冰上 30 min,42 ℃ 热激 45 s 后再放置于冰上静置 3 min。
在超净工作台中加入无抗 LB 肉汤培养基 500 μL,轻轻混匀放置于 37 ℃ 摇床中 200 rpm 震荡培养 1 h 后,5000 rpm 离心 5 min 收菌,留约 100 μL 上清轻轻吹打混匀,均匀涂布接种于含抗生素的固体 LB 平板中,置于 37 ℃ 培养箱中正放 5 min 使液体被吸收。
然后再倒置固体 LB 平板,继续在 37 ℃ 培养箱中培养 12~16 h 左右可出现单个菌落。
(5)质粒提取:挑取单个菌落于 1.5 mL EP 管中,振荡培养 1~2 h 后取一微升作为模板,进行菌液 PCR 验证,观察是否出现目的条带,进一步将上述验证正确的菌液,加入 5~20 mL 含抗生素的 LB 肉汤培养基,37 ℃ 摇床培养过夜(12~16 h),按照质粒抽提试剂盒进行提取,可以再次酶切或者测序验证。
注意事项
(1)若 PCR 产物不立即回收,应取出放在 4 ℃ 保存(短时间),或者也可琼糖凝胶电泳后切下目的片段条带,贮存于 -20 ℃(可放置时间较长),一般不建议 PCR 产物放置过久,最好直接电泳和回收。
(2)酶切时体系中的酶的用量原则是酶的总量不能超过体系的 1/10,一般在 37 ℃ 酶切 3 h 便足够。
双酶切的时候要注意同尾酶的情况,如 SalⅠ与 XhoⅠ,Bgl Ⅱ 与 BamHⅠ互为同尾酶,能产生相同的粘性末端;载体单酶切是通常要考虑载体的自连,因此连接前可以经过小牛肠碱性磷酸酶(CIP)去磷酸化处理。
常见问题
阳性克隆少。可能载体酶切不够充分,或者质粒转化感受态的效率低。可以用酶切后未连接的载体作为阴性对照,质粒作为阳性对照进行转化。



相关文章

