
验证候选基因以筛查表型
简介
考虑到筛选过程可能很复杂,并且分析产生了一个候选基因的排序列表, 有必要验证已识别的候选基因对表型的干扰。为了验证,每个针对候选基因的 sgRNA 都可以单独克隆到 sgRNA 文库的质粒主链中,并验证其筛选表型。此外,还可以量化每个 sgRNA 诱导的干扰,分別用于敲除和激活筛选的插入缺失和转录激活率,以建立表型与基因型的关系。
材料与仪器
器材:离心管、离心机等。
试剂:
① Quick-gDNA MidiPrep(Zymo Research, cat. no. D3l00)
② DNA 结合缓冲液(Zymo Research, cat. no. D4004-1-L)
③ DNA 洗涤缓冲液(Zymo Research, cat. no. D4003-2-24)
④ DNA 洗脱缓冲液(Zymo Research, cat. no. D3004-4-4)
⑤ 纯乙醇(Sigma-Aldrich, cat. no. 459844?500 mL)
⑥ 用于克隆验证 sgRNA 的引物(Integrated DNAtechnologies)
⑦ T4 多核昔酸激酶(New England BioLabs, cat. no. M0201S)
⑧ T4 DNA 连接酶反应缓冲液,10X(New England BioLabs, cat. no. B0202S)
⑨ T7 DNA 连接酶 2X 快速连接缓冲液(Enzymatics, cat. no. L6020L)
⑩ 牛血清蛋白,分子生物学等级(New England BioLabs, cat. no. B9000S)
⑪ QuickExtract DNA 提取溶液(Epicentre, cat. no. QE09050)
⑫ RNase AWAY(VWR, cat. no. 53225-514)
⑬ 蛋白酶 K(Sigma-Aldrich, cat. no. P2308-25 mg)
⑭ Tris. Imol/L, pH 8.0(Thermo Fisher, cat. no. AM9855 G)
⑮ Deoxyribonuclease I bovine(Sigma-Aldrich, cat. no. D2821-50KU)
⑯ UltraPure 1 mol/L Tris-HCl 缓冲液,pH 7.5(Thermo Fisher, cat. no. 15567027)
⑰ 氯化钙溶液(Sigma-Aldrich, cat. no. 21115-lmD 甘油(Sigma-Aldrich, cat. no. G5516-100 mL)
⑱ MgCl2, 1 mol/L(Thermo Fisher, cat. no. AM9530 G)
⑲ Triton X-l 14(Sigma-Aldrich, cat. no. XI14-1 OOmL)
⑳ 蛋白酶 K 抑制剂(EMD Millipore, cat. no. 539470-10 mg)
21. 二甲亚飒(Sigma-Aldrich, cat. no. D8418?50 mL)
22. 乙二醇■双(2-氨基乙基醍)-N, N, N, Nf -四乙酸(SigmaAldrich, cat. no. E3889-10 g)
23. Revert A id RT 逆转录试剂盒(Thermo Fisher, cat. no. K1691)
24. Oligo dT(TTTTTTTTTTTTTTTTTTTTNN; Integrated DNA Technologies)
25. TaqMan 目标探针,FAM 染料(Thermo Fisher)
26. Taqman 内源性对照探针,VIC 染料(如 Human GAPD, GAPDH, 内源 对照 VIC?/MGB 探针,弓物限制性;Thermo Fisher, cat. no. 4326317E)
27. TaqMan Fast Advanced Master Mix, 2 X(Thermo Fisher, cat. no. 4444557)
步骤
验证候选基因以筛查表型的基本过程可分为如下几步:
A. 将验证 sgRNA 克隆到 sgRNA 文库的质粒主链中。设计上链和下链引物,用于将每个候选基因的前 3 个 sgRNA 分别克隆到 sgRNA 文库的质粒主链中。克隆 2 个非靶向 sgRNA(NT1 和 NT2)进行引物对照。
B. 重悬上链和下链引物至终浓度为 100 μmol/L。准备以下混合物(表 9.20 所示),以使每种验证 sgRNA 的上游和下游引物磷酸化和退火。
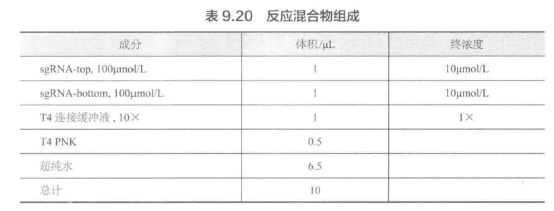
C. 使用以下条件在热循环仪中对引物进行磷酸化和退火:37 ℃ 持续 30 min :95 ℃, 5 min;以 5 ℃/min 梯度降至 25 ℃。
D. 退火反应完成后,通过添加 90 μL 超纯水以 1:10 的比例稀释磷酸化和退火的 Oligos。退火的 Oligos 可以在 -20 °C 下保存至少 1 周。
E. 通过为每个 sgRNA 设置 Golden Gate 组装反应,克隆退火的 sgRNA 插入到 sgRNA 库主链中。当克隆许多 sgRNA 时,Golden Gate 组装是有效的, 并且克隆成功率很高。按表 9.21 所示混合各组分。
关键步骤:推荐使用 FastDigest Esp3 I(Fermentas),因为其他供应商提供的 Esp3 I 的 Golden Gate 组装效率并不高。不必进行阴性对照(无插入)的 Golden Gate 组装反应,因为它始终包含菌落,因此不是克隆成功的好指标。

F. 使用以下循环条件(表 9.22)进行 Golden Gate 组装反应。完成的 Golden Gate 组装反应可在 -20 ℃ 的温度下保存至少 1 周。

G. 转化和质粒提取。将 Golden Gate 组装反应物转化至感受态大肠埃希菌菌株。推荐使用 Stbl3 菌株进行快速转化。在冰上融化化学法感受态 Stbl3 细胞,将 2 μL 来自步骤 F 的产物加到用冰预冷的 Stbl3 细胞中,并将混合物 在冰上孵育 5 min。将混合物在 42 ℃ 加热振荡 30s, 立即放冋冰中 2 min。加入 100 μL SOC 培养基,并将其铺在标准 LB 琼脂平板上(100 mm 培养皿,氨苄青霉素)。将其在 37 ℃ 下孵育过夜。
H. 第二天检查平板中菌落的生长。通常每个平板上应有数十至数百个菌落。
I. 从每个平板中,选择 1~2 个菌落进行质粒提取,以检查 sgRNA 是否正确插入以用于下游慢病毒的产生。要制备质粒提取的起始培养物,使用无 菌移液器吸头将单个菌落接种到含有 100 μg/mL 氨节青霉素的 3 mL LB 培养基 中。孵育起始培养物,并在 37℃ 以>250r/min 摇动 4?6 小时。
J. 通过将起始培养物转移到 2 个分别含有 25 mL LB 培养物(含 100 μg/mL 氨苄青霉素)的 50 mL 离心管中来扩增每个起始培养物。取下盖子,并用 AirPore 胶带密封管的顶部。孵育培养物,并于 37 ℃ 在 > 250 r/min 下摇动过夜。
K. 将起始培养物接种 12~16 小时后,使用无内毒素的试剂盒(例如 Macherey-Nagel NucleoBond Xtra Midi EF 试剂盒)提取质粒。关键步骤:使用无内毒素的质粒纯化试剂盒对于避免病毒制备和哺乳动物细胞培养中的内毒素至关重要。根据经验,用无内毒素的试剂盒制备的质粒具有更高的纯度,并能产生更高的慢病毒滴度。验证的 sgRNA 构建体可以在 -20 ℃ 下保存至少 1 年。
L. sgRNA 克隆的测序验证。通过使用 U6-fwd 引物从 U6 启动子进行测 序,验证 sgRNA 是否正确插入。将测序结果与 sgRNA 文库质粒序列进行比对,以检查 20 nt sgRNA 目标序列是否正确插入 U6 启动子和 sgRNA 支架的其余部分之间。
M. 验证细胞系的生成。通过降低慢病毒产量准备慢病毒,加入 T225 瓶或 6 孔板的 2 孔,以进行验证。使用 5 mL 注射器和 0.45 μm 注射器过滤器过滤慢病毒上清液。
N. 确定慢病毒滴度。如果同时在同一质粒主链中制备多个验证的 sgRNA 慢病毒,则滴定 2~3 种不同的 sgRNA 慢病毒,并将平均滴度扩展至其余慢病毒。
O. 与筛选过程中相似,用 MOI < 0.5 的验证 sgRNA 慢病毒转导初始细胞或表达 Cas9 成分的细胞。对于敲除验证,则选择第 7 天,以便有足够的时间形成充分的插入缺失。
P. 验证候选基因以筛选表型。一旦用于验证细胞系的抗生素选择完成之后,就可以通过筛选经验证的细胞系,并通过分析阳性、阴性和标记基因来评估细胞增殖、死亡或荧光反应,以验证筛选表型。另外,还可以确定敲除筛选的插入缺失率(步骤R)或激活筛选的倍数(步骤 S)。
R. 用于验证敲除筛选的插入缺失率分析。
1. 可以用一个两步的 PCR 方法,其中第一步使用自定义引物扩增感兴趣的基因组区域,第二步使用通用条形时引物对同一时期多达 96 个不同样本进行多重W代测序。对于每一个验证的 sgRNA, 设计自定义第1轮 NGS 引物(NGS-indel-Rl),可扩增以 sgRNA 切割位点为中心的 100~300 bp区域。设计距离靶标切割位点至少 50 bp 的引物 很重要,以允许检测更长的插入/缺失。目标退火温度为 60 ℃,使用 Primer- BLAST 检查潜在的脱靶位点。如有必要,包含 1~10 bp 的交错区域以增加文库的多样性。
2. 从验证细胞系中收集 gDNA。将验证细胞以 60% 的密度接种到 3 个 bioreps 中,置于底部黑色透明的 96 孔组织培养板中。
3. 接种 1 d 后,当细胞达到融合时,吸出培养基并加入 50 μL QuickExtract DNA 提取液。在室温下孵育 2~3 min。
4. 用移液器吸头刮下细胞,通过上下吹打彻底混匀,然后将混合物转移至 96 孔 PCR 板中。
5. 通过运行以下循环条件提取基因组 DNA(表 9.23)。

提取的基因组 DNA 可以在 -20 ℃ 下保存长达几个月。
6. 二代测序分析插入缺失的第一轮 PCR 。在以下反应中(表 9.24)使用自定义的 NGS-indel-Rl 引物,扩增每个验证和对照细胞系各自的靶区域。

关键步骤:为了使扩增 sgRNA 中的错误最小化,使用高保真聚合酶非常重要。其他高保真聚合酶,例如 PfuUltra 11(Agilent)或 NEBNext(New England BioLabs),也可以用作替代品。
7. 使用表 9.25 所示条件进行PCR循环。

8. NGS 分析插入缺失的第二轮 PCR。在以下反应中(表 9.26 所示),使 用不同的 NGS-indel-R2 引物扩增产物,对二代测序的第一轮 PCR 产物条形码化。

9. 使用与步骤7相同的循环条件进行 PCRO
10. 反应完成后,在凝胶中加入 5 μL每个扩增的靶标进行电泳,以验证单个产物是否以合适的大小成功扩增:在含有 SYBR 染料的 TBE 缓冲液中浇铸 20 g/L 琼脂糖凝胶,并在 15 V/cm 的条件下运行 30 min。
11. 混合 PCR 产物,并使用 QIAquick PCR 纯化试剂盒纯化合并的产物。关键步骤:由于 PCR 效率和产物长度不同,没有标准化的混合可能会导致二代测序表达的变化。如果不进行归一化而混合,则在测序过程中每个sgRNA 的目标是 20000~40000 个读长。或者,如果测序读长有限,请考虑分别纯化每种带条形码的 PCR 产物,通过 NanoDrop 进行定量,然后在混合之前将 PCR 产物归一化至相同浓度。
12. 如步骤 10 所述,在20 g/L 琼脂糖凝胶上电泳混合的 PCR 产物,并使用 QIAquick 凝胶提取试剂盒来提取短胶中适当大小的条带。凝胶提取的产品可以在 -20 ℃ 下保存几个月。
13. 根据 Illumina 用户手册,在 Illumina MiSeq 上对凝胶提取的样品进行测序,read 1 运行 260 个循环,index 1 运行 8 个循环,index 2 运行 8 个循环。 推荐每个 sgRNA > 10000 读长。
14. 使用 calculate indel.py 验证 sgRNA 的插入/缺失。一个 python 脚本可以用于分析二代测序结果的插入率。安装 python 2.7(https://www.python.org/ downloads/), biopython(http:// biopython.org/DIST /docs/install/Installation. html)和 SciPy(<https://www.scipy.org/install.html>)构造一个样本表,每行对应一个单独的样本。每行应在左至右各列中包含样品名称,fasta 格式或 fastq 格式文件名,引导序列,PCR 靶扩増了以及实验或对照。仅当执行最大似然估计(MLE)校正时才需要最后一列。进行 MLE 时,反映背景插入缺失率的对照样品应标记为「对照」,而实验样品应标记为「实验二请参考表 9.27 以获取样本表的示例。

15. 如果使用一个命令处理所有文件,请使用表 9.28 所示可选参数运行

为了处理单个样本,例如在并行化的清况下,请运行 pythoncaiculate_indeLpy- sample <sample name> 以生成文件 <sample name>_out.csvQ 通过调用 python compute indel.py —combine 来组合各个示例文件。运行 calculate_indel.py 后, 计算出的插入/删除将位于输出文件中,该文件还包含了完全匹配、未能对 齐,或由于质量原因而不合格或碱果替换的读长。还有三列对应于经 MLE 校正的插入缺失率,以及插入缺失 95% 置信区间的上限和下限。
S. 确定激活倍数以用于验证激活筛选。
1. 在 96 孔多聚赖氨酸包被的组织培 养板中以 60% 的融合度接种 4 个生物学重复的验证细胞,以制备细胞。
2. 反转录为 cDNA。接种约 1 天后,当细胞融合时,制备表 9.29 和表 9.30所示混合物。


除 Oligo dT 外,所有组分均可在 Thermo RevertAid RT 逆转录试剂盒中找到。 关键步骤:确保使用 RNA 时所有试剂均不含 RNA 酶,并采取适当的预防措施。
T. 将 20 μL 的逆转录混合物等分到 96 孔 PCR 板的每个孔中。解冻 RNA 裂解终止溶液,准备冷的 DPBS, 并将所有试剂(除了 RNA 完全裂解缓冲液 外)保存在冰上。
O. 从步骤 S1 的 96孔多聚赖氨酸组织培养板的每个孔中吸出的培养基,用 100 μL 冷 DPBS 洗涤,并加入 100 μL 步骤 S2)的室温 RNA 完全裂解缓冲液中。在室温下孵育,同时充分混合 6~12 min 以裂解细胞。关键步骤:将裂解时间限制在 12 min 以内,以防止 RNA 降解。
P 将 30 μL 细胞裂解液转移至新的 96 孔 PCR 板中。加入 3 μL RNA 裂解终止液以终止裂解并充分混合。带仃 RNA 裂解终止液的细胞裂解液可以保存 在 -20 °C 以进行其他逆转录反应。
Q. 然后,将 5 μL 含有 RNA 裂解终止液的细胞裂解液添加到步骤 ⑩)的逆 转录混合物中,使其总体积为 25 μL 并充分混合。
R. 在表9.31 所示循环条件下将收集的 RNA 反转录为 cDNA。

cDNA可以在 -20 ℃ 下稳定储存 1 年。
S. 使用 TaqMan qPCR 进行激活倍数分析。Thermo Fisher Scientific 已经为候选基因以及内源性对照基因(如 GAPDH 或 ACTB)提供了设计好的TaqMan 基因表达检测方法。确保实验和对照基因表达测定使用不同的探针染 料(VIC 和 FAM 染料),允许在同一反应中运行分析。为每个逆转录反应准 备以下 qPCR 预混液(表 9.32)。建议先将 TaqMan Fast Advanced Mastermix 预混合,并对所有具有相同靶基因的样本进行基因表达检测。
T. 将 4X 5 μL qPCR 预混液等分到 384 孔光学板中以进行技术重复。
U. 在表 9.33 所示循环条件下进行 qPCR
V. 一旦完成 qPCR, 使用 ddCt 方法计算候选基因相对于对照的表达倍数变化。
W. 使用死 sgRNA(dRNAs)来组合敲除和激活筛选候选基因的其他步骤。dRNAs 是具有 14 个或 15 个核昔酸间隔序列的 sgRNA, 是标准 sgRNA(具有 20 个核昔酸间隔序列)的截短版本,它仍然能够结合 DNAO dRNA 被认为是功能的」死亡「,因为它们可以引导野生型 Cas9 但不会引起双链断裂。将 MS2 结合环添加到 dRNA 主链上,可使野生型 Cas9 激活转录而无需切割。


相关文章

