
利用 PCR 进行多位点的体外基因定点突变
简介
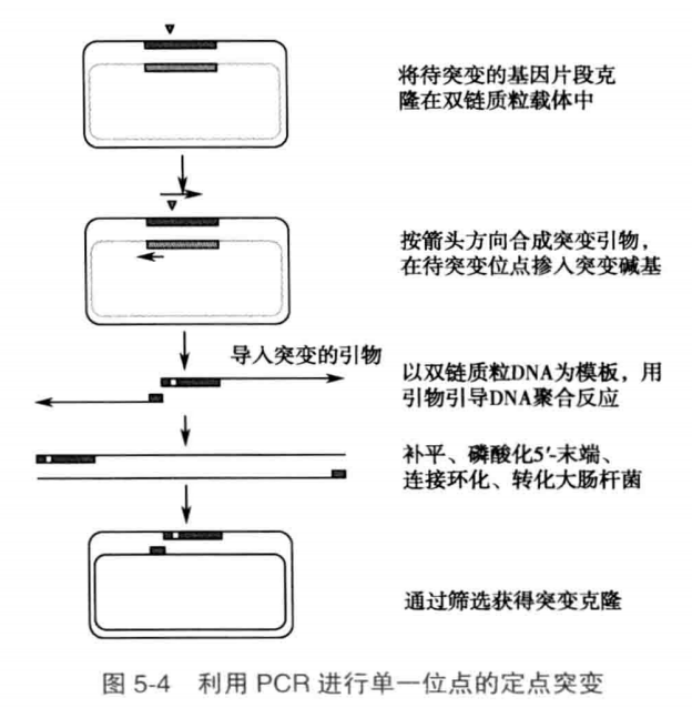
在蛋白质结构功能研究中,有时需要在多个氨基酸位点同时制作突变。以往这样的突变很难实现,随着 PCR 技术的逐步发展,对 DNA 序列上多个位点同时突变成为可能。原理利用 PCR 进行多位点的体外基因定点突变的基本原理是利用 PCR 的引物设计灵活性,在扩增中直接引入突变。
通过设计引物导入突变位点,和高保真 Taq DNA 聚合酶,以双链质粒为模板进行 PCR 反应;对 PCR 产物末端进行补平处理及 5'-端磷酸化处理;再用连接试剂进行自身连接(环化反应);然后转化、提取突变体 DNA。
材料与仪器
试剂:
① 引物
② DNA 聚合酶
③ 模板
④ 大肠杆菌感受态细胞
⑤ Dpn1 内切酶
仪器:
① PCR 仪
② 电泳仪
③ 培养箱
步骤
利用 PCR 进行多位点的体外基因定点突变的基本过程包括以下几步:
(1)用 PCR 方法合成带有多个突变位点的突变链。反应体系含超螺旋双链 DNA 模板、2 个以上含突变位点的寡核苷酸引物以及高保真 DNA 聚合酶等。
(2)采用限制性内切核酸酶 Dpn1 对 PCR 产物进行切割。Dpn1 特异识别并切割甲基化或半甲基化的位点。因模板 DNA 来自甲基化控制(dammethylaled)的菌株,所以 Dpn1 可将母代 DNA 模板切割成碎片。PCR 产物经 Dpn1 作用后,即可得到含多个突变位点的单链 DNA。
(3)将单链 DNA 转化至合适的宿主细胞,扩增为双链 DNA。提取双链的质粒 DNA,采用合适的方法进行突变鉴定。
注意事项
1、步骤(1)中需要注意的是,这里所用的寡核苷酸引物应与 DNA 的同一条模板链互补结合,由此产生的突变链既包含多个突变位点,又有一个以上缺口。缺口可由 DNA 连接酶连接。
2、含有突变位点的所有引物必须与同一条模板链退火。模板或引物的二级结构或其他因素有可能影响突变效率。如果突变效率低于 30%,可以考虑以另一条链为模板设计突变引物。
3、引物与模板结合的位置可以设计在模板的任意位置,而不影响突变效率。
4、引物的长度以 25~45 nt 为宜,Tm≥75 ℃。引物过长会导致二级结构的形成而影响与模板的结合。多条引物的 T 需要基本相等。
可用如下公式计算引物的 T:
T = 81.5 + 0.41(%G + C)- 675/N - 错配率(N:引物的长度;%G + C 和错配率取整数)。
5、突变位点应置于引物的中部,两端各有 10~15 nt 与模板互补。
6、引物的 GC 含量 ≥40%,3'-端用 1 个以上的 C 或 G 结尾。
7、每种引物在反应体系中含量应均等,引物长度相似。如果引物长度有 >;20% 的差别,需调整各引物的用量。例如,引物 1 含 25 个碱基,引物 2 含 35 个碱基,二者长度比为 1:1.4,反应体系中引物 1 与引物 2 的含量比也应为 1:1.4。


相关文章

