
培养神经元原位杂交技术实验
原理
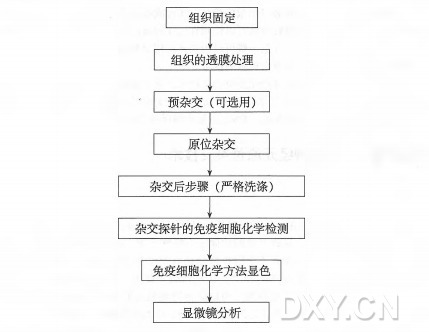
流程图
材料与仪器
RNA 聚合酶 地高辛-UTP 标记混合物
步骤
一、制备地高辛标记的探针
将 cDNAs 克隆入 pBluescript 载体中。为了能在体外转录,质粒需经线性化或 PCR 然后根据插入 cDNA 的方向应用 T3 或 T7RNA 聚合酶合成反义 RNA 探针。体外转录之后,DNA 模板用 DNA 酶 I(无 RNA 酶)降解,cDNA 水解成大约 200 个核苷酸的片段。
质粒的线性化:用合适的限制性内切酶在紧邻插入序列终止密码子的下游位点切开质粒。然后通过苯酚抽提,乙醇沉淀的方法纯化 DNA。
PCR 扩增插入序列:根据 pBluescript 中 T3 和 T7RNA 聚合酶启动子的侧翼序列设计引物,经 PCR 扩增 cDNA,然后经凝胶电泳提纯 DNA。
(这些技术的详细介绍可参见 Maniatisetal.1982)
探针制备
1.下列物质加入灭菌微量离心管:
2.短暂离心后 37℃ 温育 2 h。
3.加入 1ulDNA 酶 I(无 RNA 酶),37℃ 温育 10 min。
4.探针水解:管中加入以下物质:
5.混匀后冰浴,小于 Ikb 的探针冰浴 30 min, 大于 Ikb 的探针冰浴 60 min。
6.温至室温(室温为 25℃), 加 20 mllmol/L MES。
7.加入 28 ml7.5mol/L 乙酸铵和 412ul 冰预冷乙醇来沉淀探针(如在-70℃ 则至少沉淀 30 min, 如在-20℃ 则至少沉淀 2 h)。
8.台式离心机 12OOOr/min, 离心 10 min。
9.弃去上层清液(操作时小心勿将沉淀物倒掉),然后用 70% 的冰预冷乙醇洗涤沉淀。
10.12000r/min,离心 lOmin,尽可能将乙醇倒尽,扣干沉淀。
11.最终产物为 2~4RNA,溶于 25ul 经 DEPC 处理过水中。产物的质量浓度为 80~160ng/ul。
探针的质量控制参见第二章。
杂交缓冲液
DNA 在 90~100℃ 的 0.1~0.2md/LNa+溶液中发生变性,在低于解链温度 (Tm)25℃ 时,其复性率最高。对于原位杂交(ISH),这就意味着必须延长微量样品在 65~75℃ 中的杂交时间,然而这样的高温会严重影响细胞的形态结构。目前解决这一问题的方法主要是应用可以降低核酸双链热稳定性的有机溶剂,这样原位杂交就可以在较低的温度下进行。甲酰胺已被广泛地应用,因为在一定浓度它可以呈线性地降低 DNA 双链 Tm, 即甲酰胺的浓度每增加一个百分点,Tm 将降低 0.72℃。可以通过下面的公式计算甲酰胺对 Tm 的作用。
RNA 与 RNA 之间可以形成稳定的双链,因此,当我们利用 RNA 探针进行原位杂交时,需要严格的杂交条件。以我们的经验来看,含有 60% 甲酰胺的杂交混合物在 50℃ 杂交时,将得到很好的效果。
制备含 60% 甲酰胺的杂交混合物
在 50 ml 灭菌聚丙烯管中加入:

杂交缓冲液应避光,并在冰箱中保存。
经酸碱裂解的鲑鱼精 DNA 按下列步骤制备:
1.在 50 ml 灭菌管中加入 Ig 鲑鱼精 DNA, 加入 15 ml 经 DEPC 处理水,浸泡 15 min 到 2 h。
2.加入 2.5 ml2mol/L 盐酸,室温放置。DNA 逐渐形成白色沉淀,充分振荡直至沉淀物黏聚在一起』然后用巴士德管管尖吹打 2~3 min, 使沉淀物成球状。
3.加入 5.0 ml2.0mol/LNaOH,振荡 DNA 使之重溶,于 50℃ 温育 15 min, 以加快 DNA 溶解。
4.DEPC 处理水将混合液稀释至 175 ml, 确保无颗粒存在。
5.入 20 mlImol/LTrisHCl(pH7.4)。
6.用 2mol/L 盐酸将 DNA 溶液的 pH 调至 7.0~7.5。
用灭菌微孔滤器过滤溶液,除去所有结块沉淀:测定溶液 260nm 的吸光度。吸取 20ulDNA 溶液加入到 980ul 水中。吸光度的 40 倍即为 DNA 的质量浓度,单位为 1ug/ug。将其分装为质量浓度 4 mg/ml 的溶液,-20℃ 下保存。
培养神经元的原位杂交
神经元的分离和培养参见第十章所提供的方法。
由于我们选用 RNA 探针进行原位杂交,就必须采取措施防止 RNA 的降解。因此, 实验操作必须戴手套,使用无菌的移液器枪头、微量离心管以及不含 RNA 酶的缓冲液 [用 0.25%DEPC 处理重蒸(馏)水,37℃ 温育过夜,然后高压灭菌]。
所有的溶液都应该用孔径 0.22um 的滤器过滤,因为培养皿中的任何小颗粒都能破坏神经元。
1.细胞固定至少 2 h。应缓缓地将固定剂加入培养皿中,避免换培养液时剥离神经元细胞。固定无脊椎动物(如软体动物)神经元时,采用 1% 多聚甲醛和 1% 乙酸组成的混合液。而对于脊椎动物神经元,推荐使用 4% 多聚甲醛,乙酸的浓度可以增至 5%(Dirks1996)。乙酸是很好的核酸固定剂。
2.固定液中加入终浓度为 0.5% 的 NonadetNP40,然后温育 30 min。
3.缓冲液 1 洗涤 2 次,每次 10 min。
4.(可选用)在 37℃ 下用 0.1%~0.005% 胃蛋白酶液(溶于 0.2mol/L 盐酸中)处理细胞 10 min。此步骤可以使探针易于接近细胞中的靶 mRNA。在我们的实验中,完全省略了蛋白水解酶处理这一步,也没有发现显著的信号丢失。这样,我们省略了士骤 5~8。.
5.2% 多聚甲酵固定 4min。
6.1% 羟基氯化铵(溶于缓冲液 1 中)处理 15 min。
7.缓冲液 1 洗漆,5 min。
8.用无探针的杂交缓冲液中做预杂交,50℃ 温育 lh。此步骤不一定是必需的,但每次都应根据所选用的细胞种类核对是否需要进行预杂交。
9.用杂交缓冲液作杂交。通常 IOOul 杂交缓冲液含有 1ul 探针。杂交前,将杂交缓冲液加热至 95℃,然后立即放在冰上快速冷却。于 50℃ 孵箱温育至少 3 h, 也可温育过夜。把培养皿放入一个密闭容器中,并放置湿滤纸以防杂交缓冲液的蒸发。加入足够的杂交缓冲液以确保细胞被完全覆盖,我们通常在每个培养孔加入 100ul 缓冲液。
10.2XSSC 简单洗涤细胞 2 次,每次不超过 2 min。
11.严格洗涤。于 50℃ 用 2XSSC 和 50% 甲醜胺的混合液洗涤细胞 3 次,每次 20 min, 以去除细胞中未杂交的探针。如果背景仍然很高,可用 RNA 酶 A 处理(见步骤 12), 这样能去除所有未杂交的探针。
12.(可选用)在 2XSSC 中加入 RNA 酶 A(100ng/ul, 溶于缓冲液 1) 处理细胞 25 min。随后,进行严格洗涤(见步骤 11)。也就是说,如果选用 RNA 酶处理细胞,则需要在处理前、后各用 50% 甲酰胺和 2XSSC 混合液严格洗涤细胞 2 次。
13.2XSSC 洗漆 5 min。
14.缓冲液 1 洗漆,2X5 min。
15.TBSGT 缓冲液洗涤 15 min。
16.加入碱性磷酸酶交联的抗地高辛抗体(用 TBSGT 缓冲液 1:500 稀释),室温温育 2 h 或 4℃ 过夜。
17.缓冲液 1 洗涤 2 次,每次 5 min。
18.缓冲液 2 洗涤 2 次,每次 5 min。
19.碱性憐酸酶的底物中温育:在 1 ml 的缓冲液 2 中加入 4.5ulNBT 和 3.5ulBCIP。如神经元具有内源性碱性磷酸酶活性,加 10 一 lmol/L 左旋咪唑。每个培养皿中加入大约 0.5 ml 底物。温育时间依照细胞中转录物的量调整,最短 15 min,长可过夜,应避光温育。所用的碱性磷酸酶底物经催化生成深紫色的反应产物。如果需要荧光底物,可以使用坚固红 TR(1.0 mg/ml, 溶于缓冲液 2) 或萘酚(0.4 mg/ml,溶于缓冲液 2),通过荧光显微镜的罗丹明滤光片可以观察到突光。
20.裸眼观察到结果后,用 Tris-EDTA 缓冲液处理 15 min 以终止反应。
21.用重蒸(馏)水漂洗细胞。
22.用封片剂 aquamount 或甘油封片。
23.显微镜下观察细胞
结果
我们利用原位杂交技术来研究编码神经肽的 mRNA 在原代培养的椎实螺属动物神经元的神经突起中的分布情况。我们感兴趣的问题是转录物究竟是均匀分布在整个神经突起中,还是集聚在某一特定微小区域中。原位杂交实验显示该mRNA 在生长锥和曲张体处特别丰富(图 3-1)
常见问题
具体材料
注意:所有的材料不应含有 RNA 酶,尽可能使用厂家已灭菌的塑料制品








相关文章

