
通过PCR扩增在扩增DNA产物末端引入限制性核酸内切酶酶切位点
原理材料与仪器噬菌体 T4 DNA 连接酶 限制性内切核酸酶 靶 DNA氯仿 EDTA 乙醇 酚 氯仿 乙酸钠 TE琼脂糖凝胶 水浴箱步骤一、材料1. 缓冲液与
原理
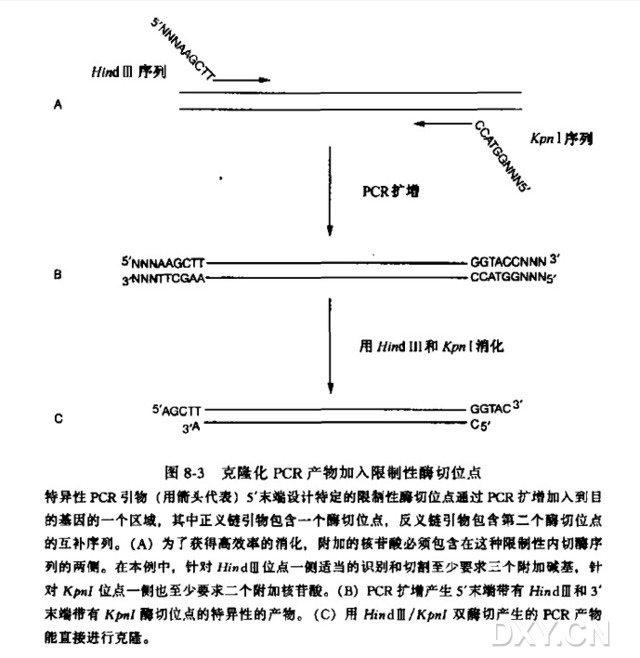
材料与仪器
噬菌体 T4 DNA 连接酶 限制性内切核酸酶 靶 DNA
氯仿 EDTA 乙醇 酚 氯仿 乙酸钠 TE
琼脂糖凝胶 水浴箱
氯仿 EDTA 乙醇 酚 氯仿 乙酸钠 TE
琼脂糖凝胶 水浴箱
步骤
一、材料
1. 缓冲液与溶液
氯仿
EDTA ( 0.5 mol/L, pH 8.0)
乙醇
酚:氯仿(1:1,V/V)
乙酸钠(3 mol/L, pH 5.2)
TE ( pH 7.5)
2. 酶与缓冲液
噬菌体 T4 DNA 连接酶
限制性内切核酸酶
3. 凝胶
琼脂糖凝胶
4. 核酸与寡核苷酸
正向引物(20 μmol/L)与反向引物(20 μmol/L)溶于水中
靶 DNA
5. 载体
质粒 DNA 应用相应的限制性内切核酸酶消化和凝胶电泳纯化
6. 特殊设备
水浴箱
二、方法
1. 应用本方案的材料部分设计的正向与反向引物,分成 4 支相同的 50 μl 体积反应管进行 PCR 反应扩增靶片段。合并 4 个反应管的反应液,溶液中应含有 200~500 ng 的期望的扩增产物。
2. 如果 PCR 产物电泳检査含有多于一或两条扩增 DNA 条带,可通过低融点琼脂糖凝胶纯化扩增片段。如果不用凝胶电泳纯化,制备的 PCR 扩增 DNA 可用酚: 氯仿抽提,和 Cemricon-100 浓缩器超滤纯化。纯化后的扩增产物溶于 TE 中(pH 7.5),浓度为 25 μg/ml。
3. 取约 100 ng 纯化后的 PCR 产物,在 20 μl 酶切反应体系中加入 1.0~2.0 单位的相应限制性内切核酸酶进行酶切消化。将反应液孵育在最适温度消化 1 h。
4. 消化结束后,用 H2O 调整反应混合液至 10 μl,加入 0.5 mol/L EDTA 至终浓度为 0.5 mol/L。将反应液用酚: 氯仿与氯仿各抽提一次。
5. 转移反应液上层水相至一个新的离心管中,加入 3 moI/L 乙酸钠(pH 5.2) 至终浓度为 0.3 mol/L,加入 2 倍体积的乙醇,在 0℃ 贮存 30 min。
6. 将离心管放置在微量离心机上 4℃ 高速离心 5 min,使 DNA 沉淀,弃上淸,然后用 70% 乙醇洗涤沉淀,溶液再离心,弃上清,将 DNA 沉淀物在空气中晾干数分钟。
7. 将 DNA 样品溶于 10 μl 水中。
8. 在一只微量离心管内,依次加入下列试剂,混合:
25 μg/ml 扩增靶 DNA 1.0 μl
质粒 DNA 20 ng
10X 连接缓冲液 1.0 μl
T4 DNA 连接酶 1 单位
用 H2O 调整终反应体积为 10 μl 反应体系。
如果需要,可加入 ATP 至终浓度 1 mmol/L。
在定向克隆的连接反应液中,纯化后靶 DNA 与酶切后的质粒载体的摩尔比例应为 1:1。设置对照反应管,加入以上除了质粒 DNA 的所有试剂。
9. 将连接反应混合液试管在 16°C 水浴温育 4 h。
10. 将上述二支试管内的连接反应液样品各取 5 μl,用 10 μl 水稀释后,转化具有抗生素抗性的感受态大肠杆菌受体菌。将这种转化菌液涂布于含合适抗生素和 IPTG 与 X-gal 的培养基的平皿上。
11. 计算实验管与对照管在培养皿上的菌落数。挑取可能含有靶基因 DNA 插入片断的白色菌落进行培养扩增,抽提质粒 DNA,利用质粒多克隆位点内的插入外源 DNA 片段侧翼的酶切位点,用相应的限制性内切核酸酶进行酶切鉴定,也可以用菌落 PCR 予以鉴定。
在不同的实验中,蓝白斑的比例在 1:5 与 2:1 之间变动。
12. 通过琼脂糖凝胶电泳鉴定克隆 DNA 片段的大小 (利用适合的 DNA marker 分子量作对照)。
13. 利用 DNA 序列分析,限制性内切核酸酶图谱或 Southern 杂交进一步鉴定克隆的靶基因 DNA 片段的正确性。
1. 缓冲液与溶液
氯仿
EDTA ( 0.5 mol/L, pH 8.0)
乙醇
酚:氯仿(1:1,V/V)
乙酸钠(3 mol/L, pH 5.2)
TE ( pH 7.5)
2. 酶与缓冲液
噬菌体 T4 DNA 连接酶
限制性内切核酸酶
3. 凝胶
琼脂糖凝胶
4. 核酸与寡核苷酸
正向引物(20 μmol/L)与反向引物(20 μmol/L)溶于水中
靶 DNA
5. 载体
质粒 DNA 应用相应的限制性内切核酸酶消化和凝胶电泳纯化
6. 特殊设备
水浴箱
二、方法
1. 应用本方案的材料部分设计的正向与反向引物,分成 4 支相同的 50 μl 体积反应管进行 PCR 反应扩增靶片段。合并 4 个反应管的反应液,溶液中应含有 200~500 ng 的期望的扩增产物。
2. 如果 PCR 产物电泳检査含有多于一或两条扩增 DNA 条带,可通过低融点琼脂糖凝胶纯化扩增片段。如果不用凝胶电泳纯化,制备的 PCR 扩增 DNA 可用酚: 氯仿抽提,和 Cemricon-100 浓缩器超滤纯化。纯化后的扩增产物溶于 TE 中(pH 7.5),浓度为 25 μg/ml。
3. 取约 100 ng 纯化后的 PCR 产物,在 20 μl 酶切反应体系中加入 1.0~2.0 单位的相应限制性内切核酸酶进行酶切消化。将反应液孵育在最适温度消化 1 h。
4. 消化结束后,用 H2O 调整反应混合液至 10 μl,加入 0.5 mol/L EDTA 至终浓度为 0.5 mol/L。将反应液用酚: 氯仿与氯仿各抽提一次。
5. 转移反应液上层水相至一个新的离心管中,加入 3 moI/L 乙酸钠(pH 5.2) 至终浓度为 0.3 mol/L,加入 2 倍体积的乙醇,在 0℃ 贮存 30 min。
6. 将离心管放置在微量离心机上 4℃ 高速离心 5 min,使 DNA 沉淀,弃上淸,然后用 70% 乙醇洗涤沉淀,溶液再离心,弃上清,将 DNA 沉淀物在空气中晾干数分钟。
7. 将 DNA 样品溶于 10 μl 水中。
8. 在一只微量离心管内,依次加入下列试剂,混合:
25 μg/ml 扩增靶 DNA 1.0 μl
质粒 DNA 20 ng
10X 连接缓冲液 1.0 μl
T4 DNA 连接酶 1 单位
用 H2O 调整终反应体积为 10 μl 反应体系。
如果需要,可加入 ATP 至终浓度 1 mmol/L。
在定向克隆的连接反应液中,纯化后靶 DNA 与酶切后的质粒载体的摩尔比例应为 1:1。设置对照反应管,加入以上除了质粒 DNA 的所有试剂。
9. 将连接反应混合液试管在 16°C 水浴温育 4 h。
10. 将上述二支试管内的连接反应液样品各取 5 μl,用 10 μl 水稀释后,转化具有抗生素抗性的感受态大肠杆菌受体菌。将这种转化菌液涂布于含合适抗生素和 IPTG 与 X-gal 的培养基的平皿上。
11. 计算实验管与对照管在培养皿上的菌落数。挑取可能含有靶基因 DNA 插入片断的白色菌落进行培养扩增,抽提质粒 DNA,利用质粒多克隆位点内的插入外源 DNA 片段侧翼的酶切位点,用相应的限制性内切核酸酶进行酶切鉴定,也可以用菌落 PCR 予以鉴定。
在不同的实验中,蓝白斑的比例在 1:5 与 2:1 之间变动。
12. 通过琼脂糖凝胶电泳鉴定克隆 DNA 片段的大小 (利用适合的 DNA marker 分子量作对照)。
13. 利用 DNA 序列分析,限制性内切核酸酶图谱或 Southern 杂交进一步鉴定克隆的靶基因 DNA 片段的正确性。

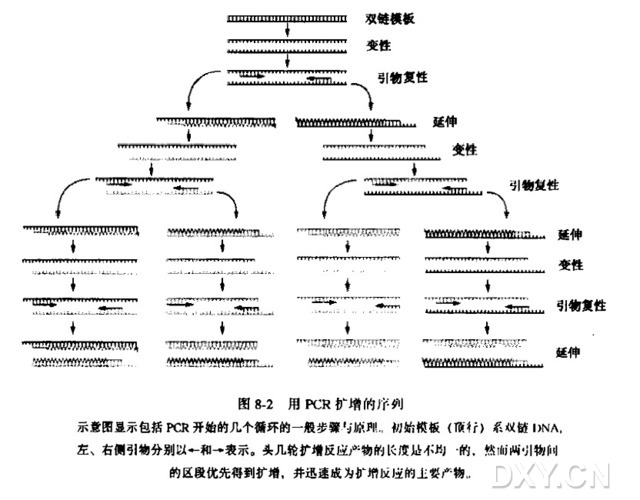
相关文章
- PCR-SSCP的发展现状
- PCR-SSCP注意的事项
- PCR-SSCP原理和操作步骤
- 内切酶在PCR反应产物中的活性(Activity of Restriction Enzymes in a Primer Extensio
- RT-PCR Analysis--详细的RT-PCR方法
- RT-PCR: The Basics
- Real-Time or Kinetic PCR
- PCR Amplification from Microbial Colonies
- Calculating Concentrations for PCR
- PURIFICATION OF PCR PRODUCTS WITH SEPHAD

