
菌落 PCR(大肠杆菌)
原理
聚合酶链反应中循环开始前的预变性和 PCR 循环过程中的变性是在 94 ℃ 或 95 ℃ 的高温下进行的,该温度可使模板 DNA 变性、双链解开,实验发现该温度也可破坏大肠杆菌菌膜,暴露感受态细胞中的 DNA 或质粒,直接挑取琼脂糖凝胶平板上的单克隆菌落可以直接用于充当 PCR 反应的模板。
用途
1、鉴别阳性克隆菌落;
2、验证重组表达载体构建是否成功;
3、扩增目的序列 DNA 片段。
材料与仪器
菌体培养平板、高压灭菌枪头、耐热的 DNA 聚合酶、 PCR 反应缓冲液、引物、dNTP、PCR 热循环仪
步骤
1、单个反应体系 PCR 混合液的制备
(1)Taq buffer(10 x)2.5 μl
(2)dNTP(2.5 mM)2 μl
(3)Primer Forward(引物浓度在 10 pmol/μl)1 μl
(4)Primer Reverse(引物浓度在 10 pmol/μl)1 μl
(5)ddH2O 18 μl
(6)Taq(2 U/μl)0.5 μl
2、用高压灭菌的 10 μl 枪头挑取菌体培养平板上的单克隆菌落,10 μl 枪头轻点菌体培养平板上的单克隆菌落即可。
3、后将 10 μl 枪头在步骤一制备的 PCR 混合液中上下晃动几下(平板上的单克隆菌落和 PCR 管均对应做好标记,如平板上单克隆菌落标记是 1#,则管子上也标 1#,以便筛选到阳性克隆后进一步扩大培养)。
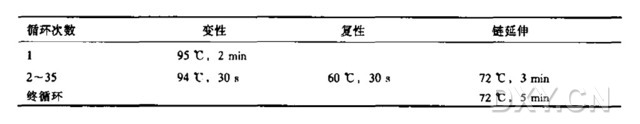
4、PCR 仪预先设置好 PCR 反应条件,根据上下游引物序列设置退火温度,根据目的序列长度和 DNA 聚合酶反应速率设置延伸时间,用于检测的 PCR 循环数可以设置在 20~25 个循环,同时设置热盖温度 105 ℃。
5、将装有 PCR 反应体系的 Tips 置于 PCR 仪中的 Tip 槽中,运行程序进行扩增。
6、程序运行完成后向各反应管中加入凝胶加样缓冲液并充分混匀,用移液枪将 DNA marker 和样品分别加入琼脂糖凝胶加样孔中跑电泳。
7、根据琼脂糖凝胶电泳条带判断阳性克隆菌落。
注意事项
1、用于质粒扩增的大肠杆菌单克隆菌落作为模板时要根据待扩增质粒是高拷贝质粒还是低拷贝质粒合理选择是否用该方法确定阳性克隆。
2、合理设计特异性引物有利于提高真阳性克隆菌落检出率。一般如果是定向克隆,用载体上的通用引物即可;如 pET 系列可用 T7 通用引物。如果是非定向克隆(如单酶切或平末端连接),一条引物用载体,一条引物用目的基因上的,这样就可以比较方便的鉴定了,而且错误概率很低。
3、使用的引物浓度不能太高,浓度过高会导致非特异性扩增,反应的循环数也不能太多,一般不超过 25 个。同时因为扩增的片段的 GC 含量问题,有的 GC 含量很低,有的又很高,导致菌落 PCR 不容易扩增出目的条带,在此建议在设置 PCR 程序时以高 GC 的温度为上限,每一循环降 0.2 度左右。
常见问题
无法筛选出阳性克隆或假阳性克隆的问题?
解决办法:高拷贝质粒的扩增菌落建议采取菌落 PCR,其它情况下需特别注意引物的特异性、Tm 值,或选取抽提质粒、稀释模板的方法筛选阳性克隆。


相关文章

