
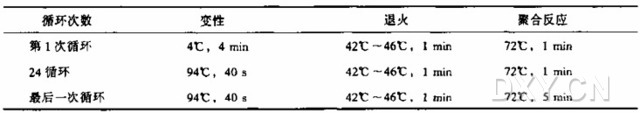
USE 诱变实验
材料与仪器带 mutS 表型(例如 BMH71-18) 的转化用大肠杆菌感受态 带 mut+表型的转化用大肠杆菌感受态退火缓冲液 贮存液 合成缓冲液 噬菌体 T
材料与仪器
带 mutS 表型(例如 BMH71-18) 的转化用大肠杆菌感受态 带 mut+表型的转化用大肠杆菌感受态
退火缓冲液 贮存液 合成缓冲液 噬菌体 T4DNA 连接酶 噬菌体 T4DNA 聚合酶或测序酶 单一位点的限制性内切核酸酶 琼脂糖凝胶 诱变引物 选择引物 质粒 DNA
70°C 水浴和适合限制酶消化反应溫度的水浴 LB 琼脂平板和含合适抗生素的 LB 液体培养基
退火缓冲液 贮存液 合成缓冲液 噬菌体 T4DNA 连接酶 噬菌体 T4DNA 聚合酶或测序酶 单一位点的限制性内切核酸酶 琼脂糖凝胶 诱变引物 选择引物 质粒 DNA
70°C 水浴和适合限制酶消化反应溫度的水浴 LB 琼脂平板和含合适抗生素的 LB 液体培养基
步骤
材料
缓冲液和溶液
贮存液,缓冲液和试剂的成分请参阅附录 1。
将贮存液稀释到合适的浓度。
10x 退火缓冲液
200 mmol/LTris-Cl(pH7.5)
100 mmol/LMgCl2
500 mmol/LNaCl
10X 合成缓冲液
100 mmol/L Tris-Cl (pH7-5)
dTTP、dATP、dCTP、dGTP 每种浓度为 5 mmol/L
10 mmol/LATP
20 mmol/LDTT
酶和缓冲液
噬菌体 T4DNA 连接酶
噬菌体 T4DNA 聚合酶或测序酶
噬菌体 T4 编码的天然 DNA 聚合酶不能从模板 DNA 上置换寡核苷酸引物 (Nossal 1974.Kunkel 1985,Bebenek and Kunkel 1989,Schens 1989)。
单一位点的限制性内切核酸酶
凝胶
1% 的琼脂糖凝胶(含 0.5ug/ml 溴化乙锭)
核酸和寡核苷酸
诱变引物
选择引物
诱变引物和选择引物一定要能退火成靶 DNA 的同一链,每条引物的 5'端一定要磷酸化。诱变引物设计见信息栏中有关诱变寡核苷酸专題。突变应位于引物的中间,突变位点两侧带有 10~15 个能与模板DNA 完全配对的碱基。寡核苷酸引物在使用前用高压液相色谱(HPLC) 或聚丙烯酰胺凝胶电泳(PAGE) 纯化(请见第 10 章,方案 1 或 5)。几个公司(Clomech;Pharmacia) 销售选择引物,某些公司(例如 Pharmacia) 也销售成对的"toggle」引物,它们可用于限制位点正向或反向转变,容许有序的多轮突变. 而不需亚克隆模板。
质粒 DNA
可通过商业销售的树脂进行柱层析或碱裂解法进行闭和环状质粒 DNA 的纯化(请见第 1 章,方案 2 或 9)。
培养基
LB 琼脂平板和含合适抗生素的 LB 液体培养基
专用设备
70°C 水浴和适合限制酶消化反应溫度的水浴
其他试剂
本方案步骤 7 和 14 需要的试剂列在第 1 章方案 23、24 或 26 中,大肠杆菌转化见方案 26。
本方案步骤 10 和 16 中少量制备质粒 DNA 需要的试剂列在第 1 章方案 1 中。
本方案步骤 17 所需的试剂列在第 12 章方案 3、4、5 中。
载体和菌株
请见附录 3
带 mutS 表型(例如,BMH71-18) 的转化用大肠杆菌感受态
带 mut+表型的转化用大肠杆菌感受态
请见步骤 14。
方法
突变 DNA 链的合成
1. 在微量离心管中混合以下物质:
10x 退火缓冲液 2ul
质粒 DNA 0.025 至 0.25pmole
选择引物 25pmoles
突变引物 25pmoles
加水至 20ul
沸水中孵育 5 min。
质粒 DNA 用量取决于反应中使用的引物和质粒 DNA。
2. 将离心管立即置于冰上冷却 5 min。微型离心机上离心 5s,使液体集中到离心管底部。
3. 将以下成分加入到含退火引物和质粒 DNA 的离心管中:
10X 合成缓冲液 3ul
噬菌体 T4DNA 聚合酶 (2~4 单位/ul) 1ul
噬菌体 T4DNA 连接酶(4~6 单位/ul) 1ul
水 5ul
用移液器温和的上下吹打几次将试剂混匀。在微型离心机上离心 5S, 使液体集中到离心管底部。37°C 孵育反应 1~2 h。
4.70°C 加热离心管至少 5 min, 灭活酶并终止反应。将离心管放置在实验台上冷却至室溫。
通过限制性内切核酸酶消化进行初筛
5. 将 NaCl 的浓度调节到适合单一位点限制酶反应的浓度。使用 10X 退火缓冲液,NaCl 贮存液,或限制酶提供的 10x 缓冲液。
在总体积为 30ul 的合成或连接混合物中,NaCl 浓度为 37.5 mmol/L。如果限制消化反应不需要 NaCl 或需要低于上述浓度的 NaCl, 可用乙醇沉淀合成连接缓冲液中的 DNA 混合物,或将 DNA 混合物通过一个悬浮在合适限制酶缓冲液中的离心柱。
6. 将 20 单位选用的限制性内切核酸酶加入到反应混合物中。在合适的溫度下消化至少 1 h。
重要:反应中的酶体积(包括聚合酶和连接酶)不应超过总反应体积的 10%。应按照以上比例相应地调节反应体积。
突变质粒的第 1 轮转化与富集
7. 按照第 1 章方案 23~26 所述的转化程序,用消化混合物中包含的质粒 DNA 转化如 BMH71-18 的 mutS 大肠杆菌菌株。
8. 用 10,50 和 250ul 转化混合物涂布含适当抗生素的 LB 琼脂平板上。37°C 孵育培养皿过夜。在培养皿孵育期间进行步骤 9。
这些培养皿被用于测定转化子的数量。每 50ul 转化混合物涂布的培养皿应产生 100~300 个克隆。
9. 加入 3 ml 含适当抗生素的 LB 培养液至剩余的转化混合物中以扩增质粒。37°C 振荡培养过夜。
10. 第二天从大约 2.5 ml 的过夜培养物中制备质粒 DNA(请见第 1 章方案 1)。
11. 用选择的限制酶消化步骤 10 制备的质粒 DNA。
质粒 DNA 500ng
10x 限制酶缓冲液 2ul
单-位点的限制性内切酶 20ul
加水 至 20ul
在适当温度中孵育反应 2 h。
12. 再加入 10 单位的限制酶,孵育至少 1h。
13. 取 5~10ul 质粒 DNA 经 1% 的琼脂糖凝胶(含 0.5ug/ml 溴化乙锭)电泳,通过凝胶电泳评估消化结果。
未被消化的(环状的)DNA 经凝胶电泳分离成两条带,它们分别对应于松弛形的环状形式或超螺旋形式 DNA。线性化的质粒 DNA 呈现与环状 DNA 不同的条带。然而,由于亲本质粒占总质粒 DNA 的绝大多数,对应于未被消化的突变体质粒 DNA 的带与被消化的亲本质粒 DNA 带相比可能相当微弱。
最后一轮转化
14. 取 2~4ul 消化的质粒 DNA(大约 50~100ng), 转化化学方法处理的大肠杆菌 mutS+菌株,或用灭菌水对 1ul 质粒 DNA 进行 5 倍稀释(约 5ng),电击法转化大肠杆菌 mutS+菌株(请见第 1 章方案 23~26)。
15. 分别取 10、50 和 250ul 转化混合物涂布含适当抗生素的 LB 琼脂平皿。37°C 孵育培养皿过夜。
16. 第 2 天,至少挑取 12 个单独的转化子进行小量质粒 DNA 制备,用限制性内切核酸酶消化,琼脂糖凝胶电泳,鉴定对抗选择性限制性内切核酸酶消化的质粒。
17.DNA 序列分析确定含预定突变的质粒(请见第 12 章方案 3、4、5)。

缓冲液和溶液
贮存液,缓冲液和试剂的成分请参阅附录 1。
将贮存液稀释到合适的浓度。
10x 退火缓冲液
200 mmol/LTris-Cl(pH7.5)
100 mmol/LMgCl2
500 mmol/LNaCl
10X 合成缓冲液
100 mmol/L Tris-Cl (pH7-5)
dTTP、dATP、dCTP、dGTP 每种浓度为 5 mmol/L
10 mmol/LATP
20 mmol/LDTT
酶和缓冲液
噬菌体 T4DNA 连接酶
噬菌体 T4DNA 聚合酶或测序酶
噬菌体 T4 编码的天然 DNA 聚合酶不能从模板 DNA 上置换寡核苷酸引物 (Nossal 1974.Kunkel 1985,Bebenek and Kunkel 1989,Schens 1989)。
单一位点的限制性内切核酸酶
凝胶
1% 的琼脂糖凝胶(含 0.5ug/ml 溴化乙锭)
核酸和寡核苷酸
诱变引物
选择引物
诱变引物和选择引物一定要能退火成靶 DNA 的同一链,每条引物的 5'端一定要磷酸化。诱变引物设计见信息栏中有关诱变寡核苷酸专題。突变应位于引物的中间,突变位点两侧带有 10~15 个能与模板DNA 完全配对的碱基。寡核苷酸引物在使用前用高压液相色谱(HPLC) 或聚丙烯酰胺凝胶电泳(PAGE) 纯化(请见第 10 章,方案 1 或 5)。几个公司(Clomech;Pharmacia) 销售选择引物,某些公司(例如 Pharmacia) 也销售成对的"toggle」引物,它们可用于限制位点正向或反向转变,容许有序的多轮突变. 而不需亚克隆模板。
质粒 DNA
可通过商业销售的树脂进行柱层析或碱裂解法进行闭和环状质粒 DNA 的纯化(请见第 1 章,方案 2 或 9)。
培养基
LB 琼脂平板和含合适抗生素的 LB 液体培养基
专用设备
70°C 水浴和适合限制酶消化反应溫度的水浴
其他试剂
本方案步骤 7 和 14 需要的试剂列在第 1 章方案 23、24 或 26 中,大肠杆菌转化见方案 26。
本方案步骤 10 和 16 中少量制备质粒 DNA 需要的试剂列在第 1 章方案 1 中。
本方案步骤 17 所需的试剂列在第 12 章方案 3、4、5 中。
载体和菌株
请见附录 3
带 mutS 表型(例如,BMH71-18) 的转化用大肠杆菌感受态
带 mut+表型的转化用大肠杆菌感受态
请见步骤 14。
方法
突变 DNA 链的合成
1. 在微量离心管中混合以下物质:
10x 退火缓冲液 2ul
质粒 DNA 0.025 至 0.25pmole
选择引物 25pmoles
突变引物 25pmoles
加水至 20ul
沸水中孵育 5 min。
质粒 DNA 用量取决于反应中使用的引物和质粒 DNA。
2. 将离心管立即置于冰上冷却 5 min。微型离心机上离心 5s,使液体集中到离心管底部。
3. 将以下成分加入到含退火引物和质粒 DNA 的离心管中:
10X 合成缓冲液 3ul
噬菌体 T4DNA 聚合酶 (2~4 单位/ul) 1ul
噬菌体 T4DNA 连接酶(4~6 单位/ul) 1ul
水 5ul
用移液器温和的上下吹打几次将试剂混匀。在微型离心机上离心 5S, 使液体集中到离心管底部。37°C 孵育反应 1~2 h。
4.70°C 加热离心管至少 5 min, 灭活酶并终止反应。将离心管放置在实验台上冷却至室溫。
通过限制性内切核酸酶消化进行初筛
5. 将 NaCl 的浓度调节到适合单一位点限制酶反应的浓度。使用 10X 退火缓冲液,NaCl 贮存液,或限制酶提供的 10x 缓冲液。
在总体积为 30ul 的合成或连接混合物中,NaCl 浓度为 37.5 mmol/L。如果限制消化反应不需要 NaCl 或需要低于上述浓度的 NaCl, 可用乙醇沉淀合成连接缓冲液中的 DNA 混合物,或将 DNA 混合物通过一个悬浮在合适限制酶缓冲液中的离心柱。
6. 将 20 单位选用的限制性内切核酸酶加入到反应混合物中。在合适的溫度下消化至少 1 h。
重要:反应中的酶体积(包括聚合酶和连接酶)不应超过总反应体积的 10%。应按照以上比例相应地调节反应体积。
突变质粒的第 1 轮转化与富集
7. 按照第 1 章方案 23~26 所述的转化程序,用消化混合物中包含的质粒 DNA 转化如 BMH71-18 的 mutS 大肠杆菌菌株。
8. 用 10,50 和 250ul 转化混合物涂布含适当抗生素的 LB 琼脂平板上。37°C 孵育培养皿过夜。在培养皿孵育期间进行步骤 9。
这些培养皿被用于测定转化子的数量。每 50ul 转化混合物涂布的培养皿应产生 100~300 个克隆。
9. 加入 3 ml 含适当抗生素的 LB 培养液至剩余的转化混合物中以扩增质粒。37°C 振荡培养过夜。
10. 第二天从大约 2.5 ml 的过夜培养物中制备质粒 DNA(请见第 1 章方案 1)。
11. 用选择的限制酶消化步骤 10 制备的质粒 DNA。
质粒 DNA 500ng
10x 限制酶缓冲液 2ul
单-位点的限制性内切酶 20ul
加水 至 20ul
在适当温度中孵育反应 2 h。
12. 再加入 10 单位的限制酶,孵育至少 1h。
13. 取 5~10ul 质粒 DNA 经 1% 的琼脂糖凝胶(含 0.5ug/ml 溴化乙锭)电泳,通过凝胶电泳评估消化结果。
未被消化的(环状的)DNA 经凝胶电泳分离成两条带,它们分别对应于松弛形的环状形式或超螺旋形式 DNA。线性化的质粒 DNA 呈现与环状 DNA 不同的条带。然而,由于亲本质粒占总质粒 DNA 的绝大多数,对应于未被消化的突变体质粒 DNA 的带与被消化的亲本质粒 DNA 带相比可能相当微弱。
最后一轮转化
14. 取 2~4ul 消化的质粒 DNA(大约 50~100ng), 转化化学方法处理的大肠杆菌 mutS+菌株,或用灭菌水对 1ul 质粒 DNA 进行 5 倍稀释(约 5ng),电击法转化大肠杆菌 mutS+菌株(请见第 1 章方案 23~26)。
15. 分别取 10、50 和 250ul 转化混合物涂布含适当抗生素的 LB 琼脂平皿。37°C 孵育培养皿过夜。
16. 第 2 天,至少挑取 12 个单独的转化子进行小量质粒 DNA 制备,用限制性内切核酸酶消化,琼脂糖凝胶电泳,鉴定对抗选择性限制性内切核酸酶消化的质粒。
17.DNA 序列分析确定含预定突变的质粒(请见第 12 章方案 3、4、5)。


相关文章
- Which housekeeping genes for qRT-PCR control-Real-Time
- What causes two peaks in melting curve-Real-Time PCR
- Detection of Viruses in Infected Plant Extracts using Immunocapture-PCR
- Inverse PCR For use with Snyder mTn-lacZ/LEU2 based mutagenesis
- Mouse p27 PCR Using Gitschier Buffer
- Real-Time PCR Quantification Using Cloned Standards and Multiple Housekeeping Genes
- Use of Internaland External Standardsor Reference RNAs for A
- WHOLE MOUNT IN SITU HYBRIDIZATION.Mouse Embryos
- RNAse A Treatment of Mouse Cells
- Natural History Museum Protocol for EST sequencing from lambda z

