
引物延伸预扩增PCR
简介
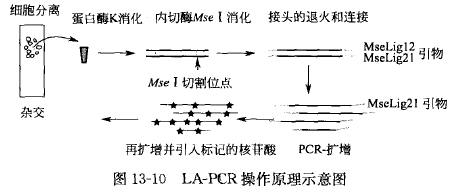
引物延伸预扩增PCR,是随机扩增整个基因组 DNA 的方法。
原理
引物延伸预扩增PCR的基本原理是PEP-PCR以随机组成的15个碱基寡核昔酸为引物,这种引物在理论上有415种排列顺序, 在低温(37°C)下随机与基因组 DNA 的大量位点退火,55°C延伸,并且每个循环的退火温度都 是由37°C连续升温至55°C,确保不同组成的引物与尽可能多的基因组序列退火,随机扩增整个 基因组 DNA。PEP-PCR可以使基因组 DNA放大几十倍。扩增产物经过特异基因位点的巢式或 半巢式PCR鉴定扩增效率和保真度。PEP-PCR的扩增效率低于DOP-PCR, 无法用常规的方法 检测产物的片段大小,但它的覆盖率却高于DOP-PCR。因而DOP-PCR用于CGH可以获得满意 的结果,而PEP-PCR的CGH信号非常微弱;但是,改良的PEP-PCR可用单个肿瘤细胞进行微 卫星的分析。
材料与仪器
器材:PCR 仪。
试剂:
①DNA模板 10μl;
②随机引物 5μl(400 Mmol/L);
③PCR 缓冲液 6μl(MgCl2 25 mmol/L, 明胶 lmg/mL , Tris-HCl 100 mmol/L, pH8. 3 ) ;
④2 mmol/L dNTP 3μl;
⑤Taq DNA 聚合酶 5U;
⑥补充水至 60μl。
步骤
引物延伸预扩增PCR的基本过程可分为如下几步:
DNA模板 10μl;随机引物 5μl(400 Mmol/L); PCR 缓冲液 6μl(MgCl2 25 mmol/L, 明胶 lmg/ml , Tris-HCl 100 mmol/L, pH8. 3 ) ; 2 mmol/L dNTP 3μl;Taq DNA 聚合酶 5U;补充水至 60μl
94°C lmin^37°C 2 min—55°C 4 min (以 l°C/10s 从 37°C升高到 55°C)共 50 个循环。
注意事项
①外源DNA和其它细胞DNA的污染是单细胞PCR的一大难题,因此需严格按操作规程操 作,避免人为污染。为减少污染风险,应建立污染评估指标,如检测多个具有高度多态性和可遗 传性的短串联重复序列(short tanderm repeat, STR)。
②PEP-PCR的重复性和扩增产物的准确性依赖于原始模板的数量和质量。由于原始模板量 少,PEP-PCR产物中每一特定DNA 序列的拷贝数也是有限的,因此在进行后续的巢式或半巢式 PCR时应考虑加入PEP-PCR产物的量。原始模板的量和 PEP-PCR产物的量决定后续PCR的质 量及结果分析。
③由于在低拷贝条件扩增容易发生错配,单细胞全基因组扩增用于植入前遗传学诊断时要 考虑到PCR 反应引入错误而导致误诊的风险,因此扩增过程中釆用高保真DNA 聚合酶是必 要的。
④对于浸润型肿瘤,肿瘤细胞常与正常细胞交织混杂,用激光显微切割有助于获得纯化的 肿瘤细胞,从而避免正常细胞的污染。
⑤PEP-PCR扩增的效率因细胞处理的条件不同而有较大的差异,福尔马林固定的石蜡切片 组织细胞的扩增效率明显低于新鲜细胞。但福尔马林固定的石蜡切片是获得纯肿瘤细胞的常用方法,因而,为获得准确的结果应尽量取更多的细胞。
⑥显微组织切割过程中容易造成染色体和DNA丢失,引起人为的等位基因丢失。而且对冰 冻或新鲜组织的处理过程要迅速,避免基因组 DNA的降解。


相关文章

