
Southern印迹技术
简介
Southern印迹技术(Southern blot)是将DNA转移到固相支持物上,然后与存在于液相中的标记探针进行杂交的过程。
Southern印迹技术可检测特定大小DNA分子的含量,常用于克隆基因的酶切图谱分析、基因组基因的定性及定量分析、基因突变分析及限制性长度多态性分析(RELP)等。
原理
Southern印迹技术的基本原理是首先将限制性核酸内切酶消化的DNA片段经凝胶电泳分离,然后使凝胶上的DNA变性,并在原位将单链DNA片段转移至硝酸纤维素膜或其他固相支持物上。
通过用放射性同位素标记特异的DNA探针,与单链DNA片段进行杂交反应,经放射性自显影,分析杂交信号,确定与探针互补的每条DNA带的位置,从而可以确定在众多酶解产物中含某一特定序列的DNA片段的位置和大小。
用途
Southern印迹技术常用于克隆基因的酶切图谱分析、基因组基因的定性及定量分析、基因突变分析及限制性长度多态性分析(RELP)等。
材料与仪器
器材:
①电泳仪、电泳槽、制胶模、梳板、玻璃板、托盘
②紫外分光光度计、紫外检测仪
③离心机、恒温水浴箱、真空烤箱
④硝酸纤维素滤膜、Whatman 3MM滤纸、吸印纸、保鲜膜、杂交袋
⑤微量移液器(20 μl)、枪头(灭菌)、1.5 ml Eppendorf 管(灭菌)
⑥放射自显影盒、X线胶片
试剂:
①材料:基因组DNA、已标记好的探针
②限制性核酸内切酶及缓冲液
③DNA分子质量标准(Marker,1kb梯度)
④琼脂糖、5 x TBE
⑤10 mg/ml EB(溴化乙锭)
⑥20 x SSC、6 x SSC、2 x SSC和0.1 x SSC
⑦50 x Denhardt's 溶液
⑧6x加样缓冲液、变性溶液、中和溶液、预杂交溶液
⑨0.1% SDS
步骤
Southern印迹的基本过程可分为如下几步:
(一)试剂配制
(1)5xTBE(Tris-硼酸-EDTA电泳缓冲液)800ml H2O溶解54.0 g Tris,27.5 g硼 酸,20 ml 0.5 mol/L EDTA(pH 8.0),再用H2O定容至1 L。
(2)6x加样缓冲液 0.25% 溴酚蓝,0.25% 二甲苯青,40% 蔗糖溶液,4 ℃保存。
(3)变性溶液 1.5 mol/L NaCl,0.5 mol/L NaOH。
(4)中和溶液 1.5 mol/L NaCl,1 mol/L Tris-HCl(pH 8.0)。
(5)20 x SSC 800 ml H2O溶解175.3 g NaCl,88.2 g柠檬酸钠,14 mol/L HC1调节 pH至7.0,用H2O定容至1 L,终浓度为3 mol/L NaCl、0.3 mol/L柠檬酸钠。
(6)6 x SSC、2 x SSC和0.1 x SSC 用20 x SSC稀释。
(7)50 x Denhardt's 溶液:1% Ficoll-400,1% PVP(聚乙烯吡咯烷酮),1% BSA(牛血清白蛋白),过滤除菌后于-20 ℃储存。
(8)预杂交溶液 6 x SSC,5 x Denhardt's 溶液,0.5% SDS,100 mg/ml鲑鱼精子DNA,50% 甲酰胺。
(9)杂交溶液 预杂交液入标记好的探针即为杂交液。
(10)20% SDS 900 ml H2O溶解200 g SDS(加热到68 ℃并用磁力搅拌器搅拌有助于溶解),浓HCl调节pH至7.2,用H2O定容至1 L,室温保存。使用时按比例稀释。
(11)EB(10 mg/ml)在100 ml水中加入1 g EB,磁力搅拌数小时,以确保其完全溶解。然后将溶液转移至棕色瓶中,保存于室温。
(12)TE 10 mmol/L Tris-HCl,1 mmol/L EDTA,pH 8.0。
(二)基因组DNA的消化
(1)按下列方法准备一个50 μl的反应体系:基因组DNA 20 μg,限制性核酸内切酶缓冲液适量,灭菌水适量,置于4 ℃数小时,期间温和地搅动DNA溶液数次,加入限制性内切酶(5U/μg,以DNA计),补足50 μl体积。
(2)4 ℃温和地搅动溶液2~3 min,再升温至酶切反应需要的温度,孵育8~12 h。
(3)消化结束后用乙醇沉淀法浓缩DNA片段,将DNA 溶于10 μl TE中,测定其OD值。
(三)琼脂糖凝胶电泳
(1)用0.5 x TBE缓冲液配制0.7% 的琼脂糖凝胶,加热使琼脂糖彻底热融,待冷却至50 ℃时,在凝胶中加入EB至终浓度为0.5 μg/ml。
(2)将融化的凝胶倒入制胶模中,梳板置于一端,底部与制胶模之间留下0.5~1 mm间隔,凝胶厚度在3~5 mm之间。
(3)待凝胶凝固后,将制胶模放入电泳槽内,并向其中加入0.5 x TBE电泳缓冲液,液面高出凝胶表面1~2 mm,小心拔出梳板。
(4)将DNA消化产物(每个加样孔不少于10 μg)与其1/5体积的6x加样缓冲液混合后,用移液器加到样品孔中。选择1kb梯度的DNA分子质量标准(Marker),将5 μl Mark-er加在凝胶的最外侧样品孔中。
(5)接通电源线,样品孔处于电泳槽的阴极端,开启电源开关。调整电压为1 V/cm,在琼脂糖凝胶中电泳12~24 h。
(6)将凝胶放在紫外灯下,观察DNA电泳条带,照相记录。
(四)转移
(1)将凝胶在加样孔一侧切去一角,作凝胶方位标记。再放进盛有变性缓冲液的盘中变性处理2次,每次15 min,轻轻摇动。
(2)将凝胶转移到中和缓冲液中中和30 min,轻轻摇动。
(3)裁1张硝酸纤维素膜,2~4张3 MM滤纸和一些吸印纸(可用卫生纸),都与胶的大小相同(硝酸纤维素膜和吸印纸不能比胶大,否则易形成旁路)。
将硝酸纤维素膜剪下一角做相应的方位记号,然后先用无菌水完全湿透,再用20 x SSC浸泡。接触胶和硝酸纤维素膜时都要戴棉布手套操作。
(4)在转移盘中放一块比胶大的平板,上面铺一张3 MM滤纸,滤纸两边浸泡在20 x SSC缓冲液中。去除滤纸与平板之间的气泡。
(5)将凝胶放置在滤纸上,使其电泳时向上的一面朝下,去除两层之间出现的气泡。然后将浸湿的硝酸纤维素膜一次准确铺在胶上,对齐。
铺膜时从一边逐渐放下,防止产生气泡,有气泡时,可用吸管赶出,不能让膜与胶下的滤纸直接接触。再用2张浸过2 x SSC缓冲液的3 MM滤纸覆盖在硝酸纤维素膜上,同样要把气泡赶去。
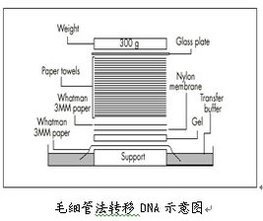
(6)把一叠干的吸印纸放置在3 MM滤纸上,在吸纸上再放一块玻璃板,加压约500 g重物(如图所示)。
(7)转移 上述步骤完毕,开始进行DNA转移。通过滤纸的吸附作用,平盘中的缓冲液就会通过胶上移,从而将DNA吸印到膜上。根据DNA复杂程度,转移2~24 h。
简单的印迹转移需2~3 h;对于基因组大片段DNA,一般需要较长时间的转移。期间如果吸印纸过于潮湿,应换新的干燥的吸印纸。
(8)固定转移后,取出硝酸纤维素膜,浸泡在6 x SSC溶液中5 min以去除琼脂糖凝胶碎块。空气干燥膜片,然后在真空烤箱内80 ℃ 烘烤2 h。烘过的膜可在室温下干燥保存,待杂交。
(五)预杂交
将转移DNA的硝酸纤维素膜放入塑料袋内,加入预杂交液(约200 μl/cm2),前后挤压塑料袋,使硝酸纤维素膜湿透。排除袋中的气泡,然后将塑料袋密封,于42 ℃水浴中孵育2~4 h,弃去预杂交液。
(六)杂交
向塑料袋中加入杂交液(预杂交液加入标记好的探针即为杂交液),重新将其密封。然后置于42 ℃水浴中杂交16~24 h。
(七)漂洗
(1)弃去杂交液,取出硝酸纤维素膜,在室温下,用2xSSC/0.1% SDS溶液漂2次,每次15 min。
(2)室温下,用0.1 x SSC/0.1% SDS溶液漂洗2次,每次 15 min。
(3)55 ℃,用0.1 x SSC/0.1% SDS溶液漂洗2次,每次30 min。
(八)放射自显影
(1)漂洗后的硝酸纤维素膜,经空气干燥,用保鲜膜包好。
(2)在暗室中,于胶片盒内在膜两侧各压上1张X线胶片,-70 ℃放射自显影。时间视杂交强度而定,24 h~10 d不等。通常曝光1~2 d后可见DNA谱带。
(九)实验结果及分析
(1)电泳结束后,应在紫外线下仔细观察DNA酶切是否完全、电泳分离效果是否良好、DNA样品有无降解、DNA带型是否清晰、有无拖尾及边缘是否模糊等现象,确认一切正常后再做转移及杂交。
注意事项
(1)EB是一种诱变剂,皮肤长期接触易发生皮肤癌。因此在操作EB时应戴上聚乙烯手套防护。
(2)DNA片段的大小决定了其转移的速度。大于15 kb的DNA片段转移时间长且转移不完全,因此对于大片段DNA的转移,可先用0.2 mol/L HC1处理15 min,脱嘌呤。但处理时间需严格掌握,过长会使DNA降解为小片段,影响转移及杂交。
(3)操作凝胶及硝酸纤维素膜时,要戴上棉布手套,以防止脏物和油脂污染,导致转移失败。
(4)不能使硝酸纤维素膜上的滤纸接触到凝胶下的滤纸,否则形成短路流动使转移不均。可用保鲜膜将凝胶边缘封围。
(5)将硝酸纤维素膜铺在凝胶上时,膜一经与凝胶接触即不可再移动。因为从接触的一刻起,转移就已经开始。
(6)杂交时,不要留过多面积,浪费预杂交液及杂交液,同时塑袋内不要留有气泡,否则预杂交及杂交都不均匀。
(7)孵育前,检查塑料袋是否密闭,勿使袋内液体流出,以及水浴箱内液体流入。
(8)漂洗强度可根据所杂交的分子大小、同源区段的大小来确定,可适当调节漂洗液强度(改变离子强度)、漂洗温度和漂洗时间,同时亦可用放射性测定仪检测漂洗情况。
(9)在杂交及放射自显影过程中,应戴上手套防护。杂交了同位素的膜片用保鲜膜包裹好,不要污染其他器皿。

相关文章

