
环状质粒 PCR 构建定点突变序列
简介
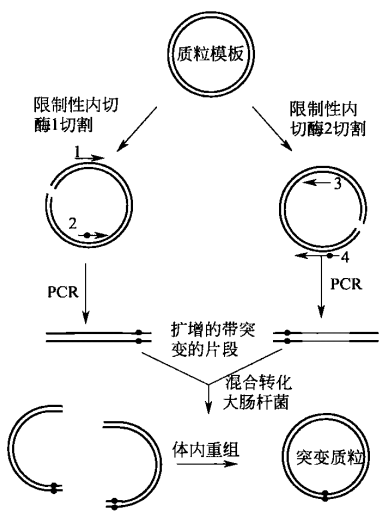
环状质粒 PCR 构建定点突变序列,是指直接将全长双链质粒 DNA 以线性形式扩增,产生一种 DNA 双链上带缺口的突变质粒,该质粒经磷酸化后自身连接形成环状闭合质粒。
原理
环状质粒 PCR 构建定点突变序列的基本原理是限制性内切酶 Dpn I 特异性切割双链 DNA 中甲基化序列 Gm6ATC。从大肠杆菌中分离的质粒已在体内的内源性 Dam 甲基化酶作用下被完全甲基化了,因而对 Dpn I 的切割敏感,半甲基化的 DNA 被 Dpn I 切割的效率较低。
相反,用四种通用脱氧核糖核苷酸体外合成的 DNA 没有甲基化,因而完全抵抗切割。在定点突变后,能用 Dpn I 降解剩余的甲基化了的野生型模板,从而富集体外合成的未甲基化 DNA。由于 Dpn I 选择性地破坏亲本模板,在 PCR 为基础的诱变中,这一方法的主要用途是净化合成的双链 DNA。
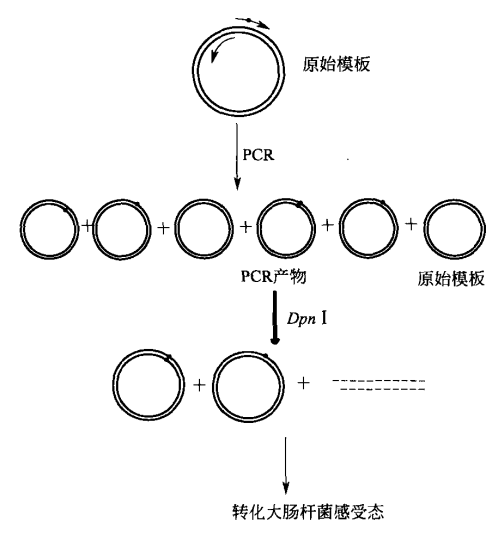
该方法以变性质粒 DNA 为模板,使用两条寡苷酸和高保真聚合酶引导 DNA 合成。经过多轮热循环后,全长双链质粒 DNA 以线性形式扩增,产生一种 DNA 双链上带缺口的突变质粒。
该质粒经磷酸化后自身连接形成环状闭合质粒。直接在 PCR 产物中或者在连接产物中加入限制性内切酶 I,切割原始质粒模板及原始模板与新合成链的杂合体,转化大肠杆菌感受态,根据突变体的复杂性和 DNA 模板长度不同,15%~80% 的转化子克隆含有需要的突变体。图解示意如下所示。
材料与仪器
器材:
PCR 扩增仪、电泳仪、水平电泳槽
试剂:
① 1 mol/L NaOH 和 1 mmol/L EDTA 溶液
② NaAc(3 mol/L,pH4.8)
③ TE(pH8.0)
④ 10 X PCR 缓 冲液:内含 15 mmol/L MgCl2,500 mmol/L KC1,100 mmol/L Tris-HCl(pH8.3,25 ℃)
⑤ 10X dNTP 混合物:内含 2 mmol/L dGTP,2 mmol/L dATP,2 mmol/L dCTP 和 2 mmol/L dTTP
⑥ 2 种寡核苷酸引物
⑦ 质粒 DNA 模板
⑧ 热稳定 DNA 聚合酶
⑨ 凝胶:含 0.5 g/ml 溴化乙锭的 1% 琼脂糖凝胶
⑩ ATP(10 mmol/L)
⑪ 噬菌体 T4 多核苷酸激酶。
⑫ 噬菌体 T4 多核苷酸激酶缓冲液:内含 10 mmol/L MgCl2 70 mmol/L Tris-HCl(25 ℃,pH7.6)、5 mmol/L DTT
⑬ 噬菌体 T4 DNA 连接酶
⑭ 噬菌体 T4 DNA 连接酶缓冲液:内含 10 mmol/L MgCl、50 mmol/L Tris-HCl(25 ℃,pH7.5)、10 mmol/L DTT、1 mmol/L ATP
⑮ Dpn I 限制性内切酶及其缓冲液
⑯ 大肠杆菌感受态
步骤
环状质粒 PCR 构建定点突变序列的基本过程可分为如下几步:
① 将 1~10 μg 质粒 DNA 溶于 40 μl 水中,加入 10 μl 1 mol/L NaOH 和 1 mmol/L EDTA 溶液。 37 ℃ 孵育变性质粒 DNA 模板。
② 加入 5 μl 3 mol/L NaAc(pH4.8)中和上述反应液,再加入 150 μl 预冷的乙醇沉淀 DNA。
③ 在微型离心机上 4 ℃ 离心 10 min 收集变性的质粒 DNA。小心弃去乙醇上清,用 150 μl 70% 乙醇清洗沉淀,重新离心 2 min 后弃上清,室温下使所有的乙醇蒸发。将 DNA 重新溶在 20 μl 水中。
④ 在一支 PCR 管中加入下列溶液混匀。10X PCR 缓冲液 10 μl、变性的质粒 DNA 模板 10 ng、热稳定 DNA 聚合酶 2 U、加水至 100 μl、10X dNTP 混合物 10 μl、两种寡核苷酸引物(10 μmol/L)各 1 μl。
⑤ PCR 扩增:13~19 次循环,每次 94 ℃ 变性 30 s,55 ℃ 退火 1 min,72 ℃ 延伸 5 min;最后两种寡核苷酸引物 (10 pmol/L) 各 1 μl,延伸 10 min。
⑥ 琼脂糖凝胶电泳检查靶 DNA 扩增情况。
⑦ 用苯酚-氯仿抽提扩增产物两次,乙醇沉淀。
⑧ 将 DNA 重新溶在 10X 噬菌体 T4 多核苷酸激酶缓冲液 10 μl,噬菌体 T4 多核苷酸激酶 10 U,10 mmol/L ATP 10 μl,加水至 100 μl,37 ℃ 孵育 1 h,68 ℃ 加热 10 min,灭活激酶。用苯酚-氯仿抽提磷酸化的 DNA 两次,乙醇沉淀收集 DNA。将上述磷酸化的 DNA 沉淀重悬在 90 μl TE 中。
⑨ 进行连接反应:10X 噬菌体 T4 DNA 连接酶缓冲液 10 μl,噬菌体 T4 DNA 连接酶 4 U,10 mmol/L ATP 10 μl,加水至 100 μl,步骤 ⑧ 磷酸化的 DNA 10~100 ng,16 ℃ 孵育 12~16 h。
⑩ 苯酚-氯仿抽提 DNA 连接产物两次,乙醇沉淀收集 DNA。DNA 沉淀重悬在 90 μl 水中。
⑪ 加入 10 μl Dpn I 缓冲液,10 U Dpn I 限制性内切酶,混匀,37 ℃ 孵育 1 h。
⑫ 消化产物转化大肠杆菌感受态。
⑬ 筛选突变体,通过序列分析进行确证。
注意事项
① 通过 PCR 构建定向突变体时为了避免碱基的错误掺入,应使用带 3'→5' 外切核酸酶校对能力的高效热稳定 DNA 聚合酶,且该酶不能具有催化非模板性掺入腺昔酸残基能力。
② 从理论上讲,模板量大将降低为获得足量产物所需的循环次数,也就减少了 Taq DNA 聚合酶诱导的碱基错误掺入的机会。然而高浓度模板有妨碍顺利扩增的倾向。另外,扩增循环次数过少会导致产生末端开放链,造成高本底。
③ 由于线性 DNA 片段转化效率低,在重组 PCR 中使用的感受态细胞转化效率要求非常高。
④ 在重组 PCR 中,如果质粒不能在 PCR 扩增部分的外侧被切开成线性,则必须纯化 PCR 产物,使其与质粒模板分开,而且如果以超螺旋质粒为模板,要进行更多次数的扩增循环,这是因为与线性模板相比,超螺旋模板 PCR 产物量要低。
⑤ 环状质粒 PCR 扩增反应混合物中 dNTP 和 Mg2+ 的起始浓度分别不要超过 250 umoI/L 和 1.5 mmol/L。
⑥ 环状质粒 PCR 线性扩增应保持在最少循环次数,单碱基置换需要 12 个循环,单个氨基酸置换需要 16 个循环,插入或缺失任何大小的 DNA 片段,使用 18 个循环,为了满足这一条件必须使用相对大量的模板 DNA。
⑦ 环状质粒 PCR 如果扩增反应正常但突变体产量低,应怀疑 Dpn I 酶的消化过程,必要时可调整 Dpn I 的用量和消化时间。
⑧ 环状质粒 PCR 如果突变效率高可不进行扩增产物的磷酸化和连接步骤,扩增产物可以直接转化大肠杆菌感受态。
⑨ 高保真热稳定 DNA 聚合酶延伸效率低于 taqDNA 聚合酶,延伸时间相对要长一些。
⑩ 少数 dam-缺陷大肠杆菌或来自其它宿主的 DNA 可用 Dam-甲基化酶进行体外甲基化。

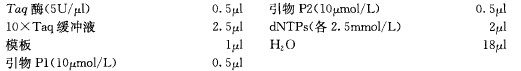
相关文章
- PCR-SSCP的发展现状
- PCR-SSCP注意的事项
- PCR-SSCP原理和操作步骤
- 内切酶在PCR反应产物中的活性(Activity of Restriction Enzymes in a Primer Extensio
- RT-PCR Analysis--详细的RT-PCR方法
- RT-PCR: The Basics
- Real-Time or Kinetic PCR
- PCR Amplification from Microbial Colonies
- Calculating Concentrations for PCR
- PURIFICATION OF PCR PRODUCTS WITH SEPHAD

